Possible cause: The unit cell may have pseudo-symmetry, or one of the cell dimensions or the space group might be given incorrectly.
Crystal class of the cell: CUBIC
Crystal class of the space group: TRICLINIC
Space group name: P 1
Atom coordinate problems and/or unexpected atoms
Note: No rounded coordinates detected
No significant rounding of atom coordinates has been detected.
Symmetry related problems
Error: Matthews Coefficient (Vm) very high
The Matthews coefficient [REF] is defined as the density of the
protein structure in cubic Angstroms per Dalton. Normal values are
between 1.5 (tightly packed, little room for solvent) and 4.0
(loosely packed, much space for solvent). Some very loosely packed
structures can get values a bit higher than that.
Numbers this high are almost always caused by giving the wrong value for Z on the CRYST1 card.
Molecular weight of all polymer chains: 16206.122
Volume of the Unit Cell V= 0.1000E+10
Cell multiplicity: 0
Matthews coefficient for observed atoms Vm= 61705.066
Nomenclature related problems
Note: Valine nomenclature OK
No errors were detected in valine nomenclature.
Note: Threonine nomenclature OK
No errors were detected in threonine nomenclature.
Note: Isoleucine nomenclature OK
No errors were detected in isoleucine nomenclature.
Note: Leucine nomenclature OK
No errors were detected in leucine nomenclature.
Note: Arginine nomenclature OK
No errors were detected in arginine nomenclature.
Note: Tyrosine torsion conventions OK
No errors were detected in tyrosine torsion angle conventions.
Note: Phenylalanine torsion conventions OK
No errors were detected in phenylalanine torsion angle conventions.
Note: Aspartic acid torsion conventions OK
No errors were detected in aspartic acid torsion angle conventions.
Note: Glutamic acid torsion conventions OK
No errors were detected in glutamic acid torsion angle conventions.
Note: Heavy atom naming OK
No errors were detected in the atom names for non-hydrogen atoms.
Warning: Chirality deviations detected
The atoms listed in the table below have an improper dihedral value
that is deviating from expected values.
Improper dihedrals are a measure of the chirality/planarity of the structure at a specific atom. Values around -35 or +35 are expected for chiral atoms, and values around 0 for planar atoms. Planar side chains are left out of the calculations, these are better handled by the planarity checks.
Three numbers are given for each atom in the table. The first is the Z-score for the improper dihedral. The second number is the measured improper dihedral. The third number is the expected value for this atom type. A final column contains an extra warning if the chirality for an atom is opposite to the expected value.
26 GLU ( 26 ) C 4.4 8.7 0.0 28 ILE ( 28 ) CA -4.4 24.7 33.8 31 SER ( 31 ) C 4.6 9.0 0.0 35 ASP ( 35 ) C 5.6 10.6 0.0 41 THR ( 41 ) C 4.3 8.4 -0.1 42 ASN ( 42 ) C 4.1 7.8 0.0 59 TYR ( 59 ) C 8.4 15.6 -0.1 63 HIS ( 63 ) C -4.6 -9.1 0.0 65 ASN ( 65 ) C 6.6 12.5 0.0 74 SER ( 74 ) C 4.6 8.9 0.0 82 GLU ( 82 ) C -4.7 -9.4 0.0 86 TYR ( 86 ) CA -4.4 24.7 34.6 91 GLN ( 91 ) C 4.9 9.3 -0.1 104 GLY ( 104 ) C 4.2 7.3 0.1 108 GLY ( 108 ) C 5.8 9.9 0.1 116 VAL ( 116 ) CA -4.4 24.0 33.6 118 GLY ( 118 ) C -4.1 -6.8 0.1 120 VAL ( 120 ) C 4.2 8.2 -0.2 121 THR ( 121 ) C 6.7 13.2 -0.1 132 ASP ( 132 ) C 5.3 10.0 0.0 138 CYS ( 138 ) C 4.2 7.8 -0.1 141 GLU ( 141 ) C 4.4 8.7 0.0
Improper dihedral RMS Z-score : 2.018
Note: Chain names are OK
All chain names assigned to polymer molecules are unique, and all
residue numbers are strictly increasing within each chain.
Note: Weights checked OK
All atomic occupancy factors ('weights') fall in the 0.0--1.0 range.
Geometric checks
Note: No missing atoms detected
All expected atoms are present.
Note: OXT check OK
All required C-terminal oxygen atoms are present.
Note: No extra C-terminal groups found
No C-terminal groups are present for non C-terminal residues
Note: All bond lengths OK
All bond lengths are in agreement with standard bond lengths using
a tolerance of 4 sigma (both standard values and sigma for amino
acid residues have been taken from Engh and Huber [REF], for
DNA/RNA from Parkinson et al [REF])
Warning: Low bond length variability
Bond lengths were found to deviate less than normal from the mean
Engh and Huber [REF] and/or Parkinson et al [REF] standard bond
lengths. The RMS Z-score given below is expected to be around 1.0
for a normally restrained data set. The fact that it is lower than
0.667 in this structure might indicate that too-strong constraints
have been used in the refinement. This can only be a problem
for high resolution X-ray structures.
RMS Z-score for bond lengths: 0.621
RMS-deviation in bond distances: 0.012
Warning: Possible cell scaling problem
Comparison of bond distances with Engh and Huber [REF] standard
values for protein residues and Parkinson et al [REF] values for
DNA/RNA shows a significant systematic deviation. It could be that
the unit cell used in refinement was not accurate enough. The
deformation matrix given below gives the deviations found: the
three numbers on the diagonal represent the relative corrections
needed along the A, B and C cell axis. These values are 1.000 in a
normal case, but have significant deviations here (significant at
the 99.99\% confidence level)
There are a number of different possible causes for the discrepancy. First the cell used in refinement can be different from the best cell calculated. Second, the value of lambda used for a synchrotron data set can be miscalibrated. Finally, the discrepancy can be caused by a dataset that has not been corrected for significant anisotropic thermal motion.
Please note that the proposed scale matrix has NOT been constrained to obey the space group symmetry. This is done on purpose. The distortions can give you an indication of the accuracy of the determination.
Unit Cell deformation matrix
| 0.998859 -0.000293 -0.000242| | -0.000293 0.996758 -0.000909| | -0.000242 -0.000909 0.996220|Proposed new scale matrix
| 0.001001 0.000000 0.000000| | 0.000000 0.001003 0.000001| | 0.000000 0.000001 0.001004|With corresponding cell
A = 998.859 B = 996.759 C = 996.220
Alpha= 90.105 Beta= 90.028 Gamma= 90.034
The CRYST1 cell dimensions
A =1000.000 B =1000.000 C =1000.000
Alpha= 90.000 Beta= 90.000 Gamma= 90.000
Variance: 39.670
(Under-)estimated Z-score: 4.642
Warning: Unusual bond angles
The bond angles listed in the table below were found to deviate
more than 4 sigma from standard bond angles (both standard values
and sigma for protein residues have been taken from Engh and Huber
[REF], for DNA/RNA from Parkinson et al [REF]). In the table below
for each strange angle the bond angle and the number of standard
deviations it differs from the standard values is given. Please
note that disulphide bridges are neglected. Atoms starting with "<"
belong to the previous residue in the sequence.
16 ASN ( 16 ) ND2 CG OD1 118.391 -4.2 17 LEU ( 17 ) C CA CB 117.806 4.1 20 PHE ( 20 ) CA CB CG 120.544 6.7 21 ASN ( 21 ) CA CB CG 117.731 5.1 35 ASP ( 35 ) CA CB CG 117.513 4.9 39 TYR ( 39 ) CB CG CD2 114.693 -4.1 41 THR ( 41 ) <C N CA 129.501 4.3 41 THR ( 41 ) CA CB CG2 117.988 4.4 46 ASP ( 46 ) CA CB CG 116.795 4.2 65 ASN ( 65 ) <C N CA 129.128 4.1 86 TYR ( 86 ) CA C O 113.551 -4.3 86 TYR ( 86 ) C CA CB 119.098 4.7 87 PRO ( 87 ) <CA <C N 125.823 5.9 87 PRO ( 87 ) N CA CB 107.403 4.0 89 HIS ( 89 ) CG ND1 CE1 109.639 4.0 105 ASP ( 105 ) CA CB CG 116.811 4.2 116 VAL ( 116 ) <CA <C N 124.909 4.1 116 VAL ( 116 ) <C N CA 135.982 7.9 116 VAL ( 116 ) N CA CB 122.762 7.2 119 ILE ( 119 ) CA CB CG1 120.328 5.8 121 THR ( 121 ) C CA CB 102.184 -4.2 126 ASN ( 126 ) CA CB CG 116.870 4.3 127 HIS ( 127 ) CA CB CG 118.551 4.8 131 ILE ( 131 ) CA CB CG1 118.108 4.5 136 PHE ( 136 ) CA CB CG 120.325 6.5
RMS Z-score for bond angles: 1.476
RMS-deviation in bond angles: 2.624
Error: Side chain planarity problems
The side chains of the residues listed in the table below contain a
planar group that was found to deviate from planarity by more than
4.0 times the expected value. For an amino acid residue that has a
side chain with a planar group, the RMS deviation of the atoms to a
least squares plane was determined. The number in the table is the
number of standard deviations this RMS value deviates from the
expected value (0.0).
76 ASP ( 76 ) 4.541 16 ASN ( 16 ) 4.315
For all atoms that are connected to an aromatic side chain in a protein residue the distance of the atom to the least squares plane through the aromatic system was determined. This value was divided by the standard deviation from a distribution of similar values from a database of small molecule structures.
127 HIS ( 127 ) CB 9.990 59 TYR ( 59 ) CB 8.950 39 TYR ( 39 ) OH 7.136 78 TYR ( 78 ) OH 6.475 14 TYR ( 14 ) OH 6.283 48 TYR ( 48 ) CB 5.763 60 TYR ( 60 ) CB 4.410 89 HIS ( 89 ) CB 4.246 78 TYR ( 78 ) CB 4.011
These scores give an impression of how ``normal'' the torsion angles in protein residues are. All torsion angles except omega are used for calculating a `normality' score. Average values and standard deviations were obtained from the residues in the WHAT IF database. These are used to calculate Z-scores. A residue with a Z-score of below -2.0 is poor, and a score of less than -3.0 is worrying. For such residues more than one torsion angle is in a highly unlikely position.
89 HIS ( 89 ) -2.8679 41 THR ( 41 ) -2.7546 116 VAL ( 116 ) -2.5762 73 THR ( 73 ) -2.4135 59 TYR ( 59 ) -2.2944 111 LEU ( 111 ) -2.2149 37 ILE ( 37 ) -2.1666 15 ARG ( 15 ) -2.1658 119 ILE ( 119 ) -2.1465 115 GLY ( 115 ) -2.1352 47 ASP ( 47 ) -2.1294 44 VAL ( 44 ) -2.1225 29 LEU ( 29 ) -2.1106 84 GLU ( 84 ) -2.0847 110 LEU ( 110 ) -2.0714 124 GLY ( 124 ) -2.0161 118 GLY ( 118 ) -2.0007
Residues with ``forbidden'' phi-psi combinations are listed, as well as residues with unusual omega angles (deviating by more than 3 sigma from the normal value). Please note that it is normal if about 5 percent of the residues is listed here as having unusual phi-psi combinations.
5 MET ( 5 ) Poor phi/psi 9 VAL ( 9 ) Poor phi/psi 26 GLU ( 26 ) omega poor 27 SER ( 27 ) omega poor 30 VAL ( 30 ) omega poor 32 TYR ( 32 ) Poor phi/psi 36 LEU ( 36 ) omega poor 41 THR ( 41 ) Poor phi/psi, omega poor 43 THR ( 43 ) omega poor 47 ASP ( 47 ) Poor phi/psi 57 ALA ( 57 ) omega poor 59 TYR ( 59 ) omega poor 60 TYR ( 60 ) omega poor 65 ASN ( 65 ) omega poor 66 ARG ( 66 ) Poor phi/psi 69 PRO ( 69 ) Poor PRO-phi 74 SER ( 74 ) Poor phi/psi 84 GLU ( 84 ) Poor phi/psi 86 TYR ( 86 ) Poor phi/psi 87 PRO ( 87 ) Poor PRO-phi 89 HIS ( 89 ) Poor phi/psi 93 ASN ( 93 ) Poor phi/psi 108 GLY ( 108 ) omega poor 115 GLY ( 115 ) omega poor 116 VAL ( 116 ) Poor phi/psi, omega poor 118 GLY ( 118 ) omega poor 125 ASP ( 125 ) Poor phi/psi 126 ASN ( 126 ) omega poor
Ramachandran Z-score : -3.205
Warning: Omega angle restraints not strong enough
The omega angles for trans-peptide bonds in a structure is
expected to give a gaussian distribution with the average around
+178 degrees, and a standard deviation around 5.5. In the current
structure the standard deviation of this distribution is above 7.0,
which indicates that the omega values have been under-constrained.
Standard deviation of omega values : 10.850
Note: chi-1/chi-2 angle correlation Z-score OK
The score expressing how well the chi-1/chi-2 angles of all residues
are corresponding to the populated areas in the database is
within expected ranges for well-refined structures.
chi-1/chi-2 correlation Z-score : -2.896
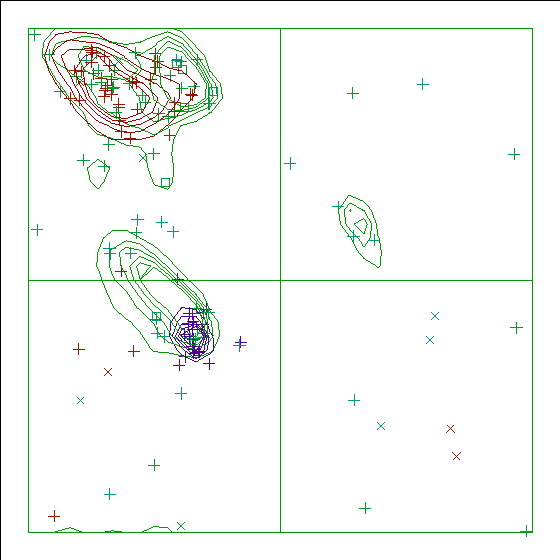
Note: Ramachandran plot
In this Ramachandran plot X-signs represent glycines, squares represent
prolines and small plus-signs represent the other residues. If too many
plus-signs fall outside the contoured areas then the molecule is poorly
refined (or worse).
In a colour picture, the residues that are part of a helix are shown in blue, strand residues in red. "Allowed" regions for helical residues are drawn in blue, for strand residues in red, and for all other residues in green.
Chain without chain identifier
Accessibility related checks
Note: Inside/Outside residue distribution normal
The distribution of residue types over the inside and the outside of the
protein is normal.
inside/outside RMS Z-score : 1.118
Note: Inside/Outside RMS Z-score plot
The Inside/Outside distribution normality RMS Z-score over a 15
residue window is plotted as function of the residue number. High
areas in the plot (above 1.5) indicate unusual inside/outside
patterns.
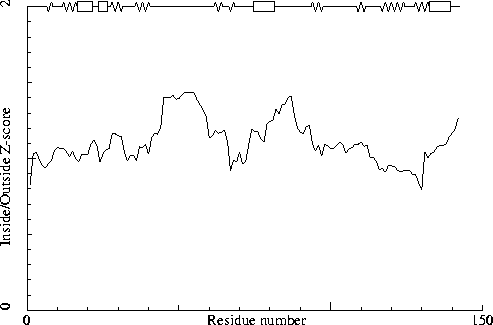
Chain without chain identifier
Secondary structure
Note: Secondary structure
This is the secondary structure according to DSSP. Only helix (H), strand
(S), turn (T) and coil (blank) are shown. [REF]
DBG> SSBOND cards to be written: 0
10 20 30 40 50 60
| | | | | |
1 - 60 GPSDMYVHVGNLIYRNLHLFNSEMHESILVSYSSDLIIYRTNTVGDDYIPSCDCTQATYY
1 - 60 SSTTTSSSSSHHHHHT 333 SSSSTTTTSSSSS T TT TT
70 80 90 100 110 120
| | | | | |
61 - 120 CKHKNRYFPITVTSHDWYEIQESEYYPKHIQYNLLIGEGPCEPGDCGGKLLCKHGVIGIV
61 - 120 SSSTTSS HHHHHHH T TTT TSSSST TT TT SSS TTT SSSS
130 140
| |
121 - 142 TAGGDNHVAFIDLRHFHCAEEQ
121 - 142 SSSSTTTSSSSSHHHHHHHTT
The contact distances of all atom pairs have been checked. Two atoms are said to `bump' if they are closer than the sum of their Van der Waals radii minus 0.40 Angstrom. For hydrogen bonded pairs a tolerance of 0.55 Angstrom is used. The first number in the table tells you how much shorter that specific contact is than the acceptable limit. The second distance is the distance between the centers of the two atoms.
The last text-item on each line represents the status of the atom pair. The text `INTRA' means that the bump is between atoms that are explicitly listed in the PDB file. `INTER' means it is an inter-symmetry bump. If the final column contains the text 'HB', the bump criterium was relaxed because there could be a hydrogen bond. Similarly relaxed criteria are used for 1--3 and 1--4 interactions (listed as 'B2' and 'B3', respectively). If the last column is 'BF', the sum of the B-factors of the atoms is higher than 80, which makes the appearance of the bump somewhat less severe because the atoms probably aren't there anyway.
Bumps between atoms for which the sum of their occupancies is lower than one are not reported. In any case, each bump is listed in only one direction.
112 CYS ( 112 ) SG -- 114 HIS ( 114 ) ND1 0.107 3.193 INTRA 5 MET ( 5 ) SD -- 107 GLY ( 107 ) O 0.096 2.904 INTRA 137 HIS ( 137 ) CG -- 138 CYS ( 138 ) N 0.079 3.021 INTRA 5 MET ( 5 ) CG -- 6 TYR ( 6 ) N 0.057 3.043 INTRA 8 HIS ( 8 ) O -- 9 VAL ( 9 ) C 0.042 2.758 INTRA 138 CYS ( 138 ) SG -- 139 ALA ( 139 ) N 0.013 3.287 INTRA 119 ILE ( 119 ) CG2 -- 131 ILE ( 131 ) C 0.001 3.199 INTRA
The packing environment of the residues is compared with the average packing environment for all residues of the same type in good PDB files. A low packing score can indicate one of several things: Poor packing, misthreading of the sequence through the density, crystal contacts, contacts with a co-factor, or the residue is part of the active site. It is not uncommon to see a few of these, but in any case this requires further inspection of the residue.
86 TYR ( 86 ) -7.52 59 TYR ( 59 ) -6.27 113 LYS ( 113 ) -6.11 92 TYR ( 92 ) -5.98 114 HIS ( 114 ) -5.24 56 GLN ( 56 ) -5.22 48 TYR ( 48 ) -5.02
The protein is probably threaded correctly, but either poorly refined, or it is just a protein with an unusual (but correct) structure. The average quality of 200 highly refined Xray structures was -0.5+/-0.4 [REF].
Average for range 1 - 142 : -1.735
Note: Quality value plot
The quality value smoothed over a 10 residue window is plotted as
function of the residue number. Low areas in the plot (below
-2.0) indicate "unusual" packing.
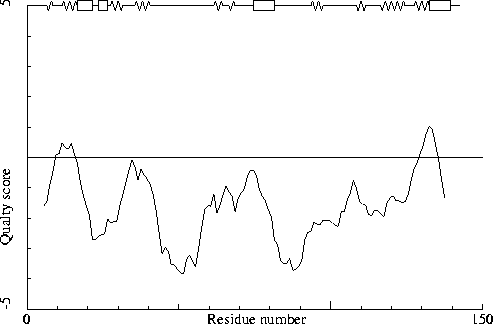
Chain without chain identifier
Warning: Low packing Z-score for some residues
The residues listed in the table below have an unusual packing
environment according to the 2nd generation quality check. The score
listed in the table is a packing normality Z-score: positive means
better than average, negative means worse than average. Only residues
scoring less than -2.50 are listed here. These are the "unusual"
residues in the structure, so it will be interesting to take a
special look at them.
25 HIS ( 25 ) -3.12 114 HIS ( 114 ) -3.05 86 TYR ( 86 ) -2.74 24 MET ( 24 ) -2.57 90 ILE ( 90 ) -2.53
A molecule is certain to be incorrect if the Z-score is below -5.0. Poorly refined molecules, very well energy minimized misthreaded molecules and low homology models give values between -2.0 and -5.0. The average quality of properly refined Xray structures is 0.0+/-1.0.
All contacts : Average = -0.798 Z-score = -5.13
BB-BB contacts : Average = -0.226 Z-score = -1.61
BB-SC contacts : Average = -0.773 Z-score = -4.16
SC-BB contacts : Average = -0.419 Z-score = -2.38
SC-SC contacts : Average = -0.927 Z-score = -4.79
Note: Second generation quality Z-score plot
The second generation quality Z-score smoothed over a 10 residue window
is plotted as function of the residue number. Low areas in the plot (below
-1.3) indicate "unusual" packing.
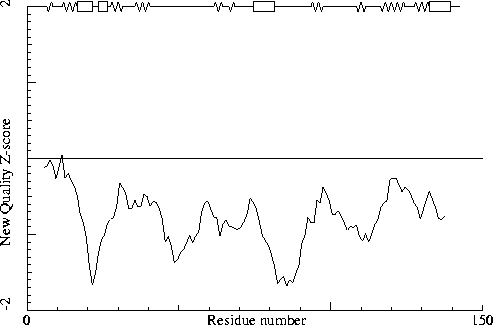
Chain without chain identifier
Warning: Backbone oxygen evaluation
The residues listed in the table below have an unusual backbone
oxygen position.
For each of the residues in the structure, a search was performed to find 5-residue stretches in the WHAT IF database with superposable C-alpha coordinates, and some constraints on the neighboring backbone oxygens.
In the following table the RMS distance between the backbone oxygen positions of these matching structures in the database and the position of the backbone oxygen atom in the current residue is given. If this number is larger than 1.5 a significant number of structures in the database show an alternative position for the backbone oxygen. If the number is larger than 2.0 most matching backbone fragments in the database have the peptide plane flipped. A manual check needs to be performed to assess whether the experimental data can support that alternative as well. The number in the last column is the number of database hits (maximum 80) used in the calculation. It is "normal" that some glycine residues show up in this list, but they are still worth checking!
124 GLY ( 124 ) 2.71 12 44 VAL ( 44 ) 1.63 17 69 PRO ( 69 ) 1.54 11
It is not necessarily an error if a few residues have rotamer values below 0.3, but careful inspection of all residues with these low values could be worth it.
31 SER ( 31 ) 0.35 38 ILE ( 38 ) 0.40
For this check, backbone conformations are compared with database structures using C-alpha superpositions with some restraints on the backbone oxygen positions.
A residue mentioned in the table can be part of a strange loop, or there might be something wrong with it or its directly surrounding residues. There are a few of these in every protein, but in any case it is worth looking at!
5 MET ( 5 ) 0 9 VAL ( 9 ) 0 24 MET ( 24 ) 0 32 TYR ( 32 ) 0 41 THR ( 41 ) 0 47 ASP ( 47 ) 0 60 TYR ( 60 ) 0 64 LYS ( 64 ) 0 65 ASN ( 65 ) 0 66 ARG ( 66 ) 0 74 SER ( 74 ) 0 85 TYR ( 85 ) 0 89 HIS ( 89 ) 0 116 VAL ( 116 ) 0 119 ILE ( 119 ) 0 125 ASP ( 125 ) 0 27 SER ( 27 ) 1 29 LEU ( 29 ) 1 73 THR ( 73 ) 1 86 TYR ( 86 ) 1 106 CYS ( 106 ) 1 122 ALA ( 122 ) 1 28 ILE ( 28 ) 2 40 ARG ( 40 ) 2 61 CYS ( 61 ) 2 114 HIS ( 114 ) 2 126 ASN ( 126 ) 2 127 HIS ( 127 ) 2
Backbone conformation Z-score : -6.189
B-factor analysis
Warning: Average B-factor problem
The average B-factor for all buried protein atoms normally lies between
10--20. Values around 3--5 are expected for X-ray studies performed
at liquid nitrogen temperature.
Because of the extreme value for the average B-factor, no further analysis of the B-factors is performed.
Average B-factor for buried atoms : 0.000
Warning: B-factor plot impossible
All average B-factors are zero. Plot suppressed.
Chain without chain identifier
Hydrogen bond related checks
Error: HIS, ASN, GLN side chain flips
Listed here are Histidine, Asparagine or Glutamine residues for
which the orientation determined from hydrogen bonding analysis are
different from the assignment given in the input. Either they could
form energetically more favorable hydrogen bonds if the terminal
group was rotated by 180 degrees, or there is no assignment in the
input file (atom type 'A') but an assignment could be made. If a
residue is marked ``flexible'' the flipped conformation is only
slightly better than the non-flipped conformation.
65 ASN ( 65 ) 81 GLN ( 81 ) 127 HIS ( 127 ) 137 HIS ( 137 )
In the table below all normal histidine residues are listed. The assignment based on the geometry of the residue is listed first, together with the RMS Z-score for the fit to the Engh and Huber parameters. For all residues where the H-bond assignment is different, the assignment is listed in the last columns, together with its RMS Z-score to the Engh and Huber parameters.
As always, the RMS Z-scores should be close to 1.0 if the residues were restrained to the Engh and Huber parameters during refinement.
Please note that because the differences between the geometries of the different types are small it is possible that the geometric assignment given here does not correspond to the type used in refinement. This is especially true if the RMS Z-scores are much higher than 1.0.
If the two assignments differ, or the ``geometry'' RMS Z-score is high, it is advisable to verify the hydrogen bond assignment, check the HIS type used during the refinement and possibly adjust it.
8 HIS ( 8 ) HIS-E 1.67 18 HIS ( 18 ) HIS-E 1.95 25 HIS ( 25 ) HIS-E 1.95 63 HIS ( 63 ) HIS-E 1.91 HIS-D 2.47 75 HIS ( 75 ) HIS-E 2.02 89 HIS ( 89 ) HIS-E 1.82 114 HIS ( 114 ) HIS-E 1.74 127 HIS ( 127 ) HIS-E 1.75 HIS-D 2.45 135 HIS ( 135 ) HIS-E 1.86 137 HIS ( 137 ) HIS-E 1.72 HIS-D 2.22
Hydrogen bond donors that are buried inside the protein normally use all of their hydrogens to form hydrogen bonds within the protein. If there are any non hydrogen bonded buried hydrogen bond donors in the structure they will be listed here. In very good structures the number of listed atoms will tend to zero.
6 TYR ( 6 ) N 8 HIS ( 8 ) NE2 16 ASN ( 16 ) N 16 ASN ( 16 ) ND2 30 VAL ( 30 ) N 44 VAL ( 44 ) N 56 GLN ( 56 ) N 65 ASN ( 65 ) ND2 74 SER ( 74 ) N 84 GLU ( 84 ) N 85 TYR ( 85 ) OH 86 TYR ( 86 ) N 90 ILE ( 90 ) N 91 GLN ( 91 ) N 91 GLN ( 91 ) NE2 92 TYR ( 92 ) N 96 ILE ( 96 ) N 99 GLY ( 99 ) N 102 GLU ( 102 ) N 106 CYS ( 106 ) N
Side-chain hydrogen bond acceptors that are buried inside the protein normally form hydrogen bonds within the protein. If there are any not hydrogen bonded in the optimized hydrogen bond network they will be listed here.
8 HIS ( 8 ) ND1 81 GLN ( 81 ) OE1 82 GLU ( 82 ) OE1 142 GLN ( 142 ) OE1
The second part of the table mostly gives an impression of how well the model conforms to common refinement constraint values. The first part of the table shows a number of constraint-independent quality indicators.
Structure Z-scores, positive is better than average:
1st generation packing quality : -3.088 2nd generation packing quality : -5.126 (bad) Ramachandran plot appearance : -3.205 (poor) chi-1/chi-2 rotamer normality : -2.896 Backbone conformation : -6.189 (bad)
Bond lengths : 0.621 (tight) Bond angles : 1.476 Omega angle restraints : 1.973 (loose) Side chain planarity : 1.600 Improper dihedral distribution : 2.018 (loose) Inside/Outside distribution : 1.118