Improper dihedral RMS Z-score : 0.577
Note: Chain names are OK
All chain names assigned to polymer molecules are unique, and all
residue numbers are strictly increasing within each chain.
Note: Weights checked OK
All atomic occupancy factors ('weights') fall in the 0.0--1.0 range.
Geometric checks
Note: No missing atoms detected
All expected atoms are present.
Warning: C-terminal oxygen atoms missing
The C-atoms listed in the table below belong to a C-terminal residue
in a protein chain, but the C-terminal oxygen ("O2" or "OXT") that it
should be bound to was not found.
145 MET ( 423 ) C
RMS Z-score for bond lengths: 0.765
RMS-deviation in bond distances: 0.017
Note: No bond length directionality
Comparison of bond distances with Engh and Huber [REF] standard
values for protein residues and Parkinson et al [REF] values for
DNA/RNA does not show significant systematic deviations.
Note: All bond angles OK
All bond angles are in agreement with standard bond angles using a
tolerance of 4 sigma (both standard values and sigma for protein
residues have been taken from Engh and Huber [REF], for DNA/RNA
from Parkinson et al. [REF]). Please note that only bond angles
within protein residues are taken into account: disulphide bridges
and peptide bonds are neglected.
Note: Normal bond angle variability
Bond angles were found to deviate normally from the mean standard
bond angles (normal values for protein residues were taken from
Engh and Huber [REF], for DNA/RNA from Parkinson et al [REF]). The
RMS Z-score given below is expected to be around 1.0 for a normally
restrained data set, and this is indeed observed for very high
resolution X-ray structures. More common values are around 1.55
RMS Z-score for bond angles: 0.913
RMS-deviation in bond angles: 1.799
Note: Side chain planarity OK
All of the side chains of residues that have a planar group are
planar within expected RMS deviations.
Note: Atoms connected to aromatic rings OK
All of the atoms that are connected to planar aromatic rings in side
chains of amino-acid residues are in the plane within expected RMS
deviations.
Note: PRO puckering amplitude OK
Puckering amplitudes for all PRO residues are within normal ranges.
Warning: Unusual PRO puckering phases
The proline residues listed in the table below have a puckering phase
that is not expected to occur in protein structures. Puckering
parameters were calculated by the method of Cremer and Pople
[REF]. Normal PRO rings approximately show a so-called envelope
conformation with the C-gamma atom above the plane of the ring
(phi=+72 degrees), or a half-chair conformation with C-gamma below
and C-beta above the plane of the ring (phi=-90 degrees). If phi
deviates strongly from these values, this is indicative of a very
strange conformation for a PRO residue, and definitely requires a
manual check of the data.
73 PRO ( 348 ) 102.1 envelop C-beta (108 degrees)
These scores give an impression of how ``normal'' the torsion angles in protein residues are. All torsion angles except omega are used for calculating a `normality' score. Average values and standard deviations were obtained from the residues in the WHAT IF database. These are used to calculate Z-scores. A residue with a Z-score of below -2.0 is poor, and a score of less than -3.0 is worrying. For such residues more than one torsion angle is in a highly unlikely position.
88 THR ( 363 ) -2.8118 73 PRO ( 348 ) -2.8071 72 PHE ( 347 ) -2.6945 108 VAL ( 385 ) -2.1320 3 ARG ( 276 ) -2.1063 116 CYS ( 393 ) -2.0680 76 GLY ( 351 ) -2.0213
Residues with ``forbidden'' phi-psi combinations are listed, as well as residues with unusual omega angles (deviating by more than 3 sigma from the normal value). Please note that it is normal if about 5 percent of the residues is listed here as having unusual phi-psi combinations.
2 GLY ( 275 ) Poor phi/psi 3 ARG ( 276 ) Poor phi/psi 6 ARG ( 279 ) Poor phi/psi 7 GLY ( 280 ) Poor phi/psi 73 PRO ( 348 ) Poor PRO-phi 74 GLY ( 349 ) Poor phi/psi 81 LEU ( 356 ) Poor phi/psi 88 THR ( 363 ) Poor phi/psi 103 ASP ( 380 ) Poor phi/psi 115 MET ( 392 ) omega poor
Ramachandran Z-score : -2.050
Warning: Omega angles too tightly restrained
The omega angles for trans-peptide bonds in a structure are
expected to give a gaussian distribution with the average around
+178 degrees and a standard deviation around 5.5 degrees. These
expected values were obtained from very accurately determined
structures. Many protein structures are too tightly constrained.
This seems to be the case with the current structure, as the
observed standard deviation is below 4.0 degrees.
Standard deviation of omega values : 3.651
Note: chi-1/chi-2 angle correlation Z-score OK
The score expressing how well the chi-1/chi-2 angles of all residues
are corresponding to the populated areas in the database is
within expected ranges for well-refined structures.
chi-1/chi-2 correlation Z-score : -0.850
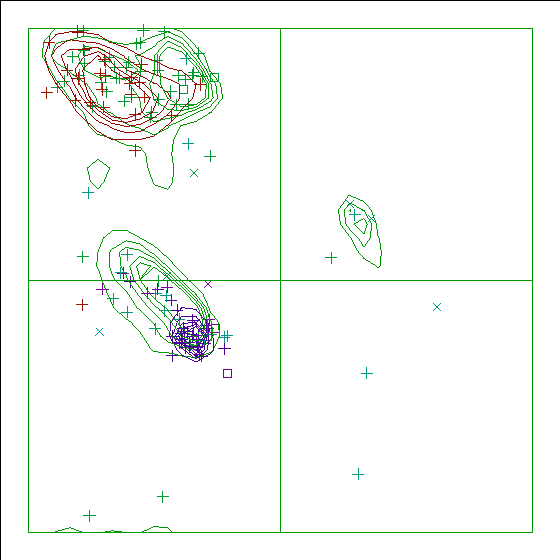
Note: Ramachandran plot
In this Ramachandran plot X-signs represent glycines, squares represent
prolines and small plus-signs represent the other residues. If too many
plus-signs fall outside the contoured areas then the molecule is poorly
refined (or worse).
In a colour picture, the residues that are part of a helix are shown in blue, strand residues in red. "Allowed" regions for helical residues are drawn in blue, for strand residues in red, and for all other residues in green.
Chain without chain identifier
Accessibility related checks
Note: Inside/Outside residue distribution normal
The distribution of residue types over the inside and the outside of the
protein is normal.
inside/outside RMS Z-score : 1.145
Note: Inside/Outside RMS Z-score plot
The Inside/Outside distribution normality RMS Z-score over a 15
residue window is plotted as function of the residue number. High
areas in the plot (above 1.5) indicate unusual inside/outside
patterns.
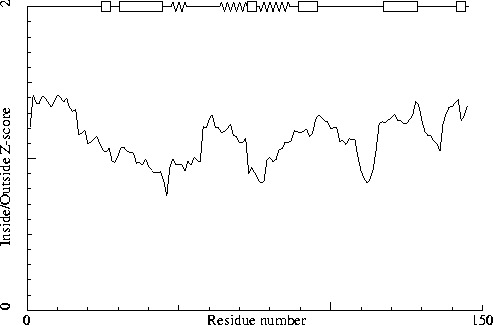
Chain without chain identifier
Secondary structure
Note: Secondary structure
This is the secondary structure according to DSSP. Only helix (H), strand
(S), turn (T) and coil (blank) are shown. [REF]
DBG> SSBOND cards to be written: 0
DBG> SSBOND cards to be written: 0
DBG> SSBOND cards to be written: 0
DBG> SSBOND cards to be written: 0
10 20
| |
1 - 29 HGRDTRGIFRVHQFEKIEQFVYTSPHDNK
1 - 29 TT T TT 333
30 40 50 60 70 80
| | | | | |
30 - 89 FEEMIGTAEEFYQSLGIPYHIVNIVSGSLNHAASKKLDLEAWFPGSGAFRELVSCSNCTD
30 - 89 HHHHHHHHHHHHHHT SSSSS TTTT TT TSSSSSSSSS333TSSSSSSSSSS TT
90 100
| |
90 - 100 YQARRLRIRYG
90 - 100 HHHHHHT
110 120 130
| | |
101 - 136 MMDKVEFVHMLNATMCATTRTICAILENYQTEKGII
101 - 136 T HHHHHHHHHHHT TTT
140
|
137 - 145 VPEKLREFM
137 - 145 TTT333
The contact distances of all atom pairs have been checked. Two atoms are said to `bump' if they are closer than the sum of their Van der Waals radii minus 0.40 Angstrom. For hydrogen bonded pairs a tolerance of 0.55 Angstrom is used. The first number in the table tells you how much shorter that specific contact is than the acceptable limit. The second distance is the distance between the centers of the two atoms.
The last text-item on each line represents the status of the atom pair. The text `INTRA' means that the bump is between atoms that are explicitly listed in the PDB file. `INTER' means it is an inter-symmetry bump. If the final column contains the text 'HB', the bump criterium was relaxed because there could be a hydrogen bond. Similarly relaxed criteria are used for 1--3 and 1--4 interactions (listed as 'B2' and 'B3', respectively). If the last column is 'BF', the sum of the B-factors of the atoms is higher than 80, which makes the appearance of the bump somewhat less severe because the atoms probably aren't there anyway.
Bumps between atoms for which the sum of their occupancies is lower than one are not reported. In any case, each bump is listed in only one direction.
84 CYS ( 359 ) SG -- 113 ALA ( 390 ) CB 0.902 2.498 INTRA 30 PHE ( 305 ) CE1 -- 84 CYS ( 359 ) O 0.375 2.425 INTRA 81 LEU ( 356 ) CD2 -- 120 ARG ( 397 ) CB 0.373 2.827 INTRA 84 CYS ( 359 ) CB -- 113 ALA ( 390 ) CB 0.367 2.833 INTRA 33 MET ( 308 ) CE -- 111 LEU ( 388 ) CB 0.352 2.848 INTRA 40 PHE ( 315 ) CD2 -- 116 CYS ( 393 ) SG 0.328 3.072 INTRA 80 GLU ( 355 ) O -- 81 LEU ( 356 ) CD2 0.310 2.490 INTRA 50 ILE ( 325 ) CG2 -- 66 LEU ( 341 ) CD2 0.258 2.942 INTRA 54 VAL ( 329 ) O -- 55 SER ( 330 ) C 0.220 2.580 INTRA 30 PHE ( 305 ) CE1 -- 34 ILE ( 309 ) CD1 0.218 2.982 INTRA 53 ILE ( 328 ) CG2 -- 57 SER ( 332 ) OG 0.215 2.585 INTRA 28 ASN ( 301 ) CG -- 109 HIS ( 386 ) ND1 0.214 2.886 INTRA 137 VAL ( 415 ) O -- 138 PRO ( 416 ) C 0.211 2.589 INTRA 88 THR ( 363 ) CG2 -- 89 ASP ( 364 ) N 0.206 2.894 INTRA 41 TYR ( 316 ) OH -- 81 LEU ( 356 ) CB 0.193 2.607 INTRA 84 CYS ( 359 ) CA -- 113 ALA ( 390 ) CB 0.187 3.013 INTRA 81 LEU ( 356 ) CD2 -- 120 ARG ( 397 ) CG 0.179 3.021 INTRA 53 ILE ( 328 ) CG2 -- 58 LEU ( 333 ) N 0.178 2.922 INTRA 86 ASN ( 361 ) ND2 -- 88 THR ( 363 ) N 0.173 2.827 INTRA 82 VAL ( 357 ) CG2 -- 116 CYS ( 393 ) CB 0.168 3.032 INTRA 53 ILE ( 328 ) CD1 -- 65 LYS ( 340 ) CG 0.160 3.040 INTRA 110 MET ( 387 ) C -- 111 LEU ( 388 ) CD2 0.158 3.042 INTRA 87 CYS ( 362 ) O -- 88 THR ( 363 ) C 0.152 2.648 INTRA 59 ASN ( 334 ) N -- 65 LYS ( 340 ) NZ 0.144 2.856 INTRA 73 PRO ( 348 ) O -- 75 SER ( 350 ) N 0.142 2.558 INTRAAnd so on for a total of 80 lines
The packing environment of the residues is compared with the average packing environment for all residues of the same type in good PDB files. A low packing score can indicate one of several things: Poor packing, misthreading of the sequence through the density, crystal contacts, contacts with a co-factor, or the residue is part of the active site. It is not uncommon to see a few of these, but in any case this requires further inspection of the residue.
6 ARG ( 279 ) -7.41 10 ARG ( 283 ) -6.77 13 GLN ( 286 ) -6.47 102 MET ( 379 ) -6.08 3 ARG ( 276 ) -5.45 132 GLU ( 409 ) -5.45 12 HIS ( 285 ) -5.39 144 PHE ( 422 ) -5.12 26 HIS ( 299 ) -5.10
Average for range 1 - 145 : -1.391
Note: Quality value plot
The quality value smoothed over a 10 residue window is plotted as
function of the residue number. Low areas in the plot (below
-2.0) indicate "unusual" packing.
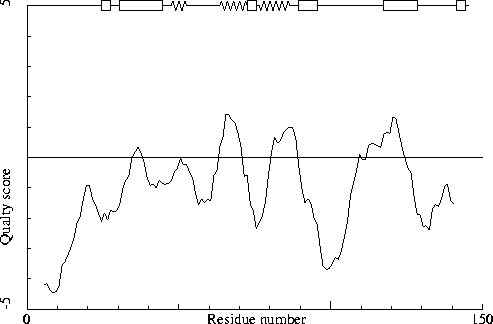
Chain without chain identifier
Warning: Low packing Z-score for some residues
The residues listed in the table below have an unusual packing
environment according to the 2nd generation quality check. The score
listed in the table is a packing normality Z-score: positive means
better than average, negative means worse than average. Only residues
scoring less than -2.50 are listed here. These are the "unusual"
residues in the structure, so it will be interesting to take a
special look at them.
9 PHE ( 282 ) -3.25 12 HIS ( 285 ) -3.12 26 HIS ( 299 ) -3.08 60 HIS ( 335 ) -2.95
The table below lists the first and last residue in each stretch found, as well as the average residue Z-score of the series.
8 ILE ( 281 ) --- 13 GLN ( 286 ) -2.46
The protein is probably threaded correctly, but either poorly refined, or it is just a protein with an unusual (but correct) structure. The average quality of properly refined Xray structures is 0.0+/-1.0.
All contacts : Average = -0.499 Z-score = -3.15
BB-BB contacts : Average = -0.017 Z-score = -0.11
BB-SC contacts : Average = -0.658 Z-score = -3.53
SC-BB contacts : Average = -0.064 Z-score = -0.22
SC-SC contacts : Average = -0.614 Z-score = -3.06
Note: Second generation quality Z-score plot
The second generation quality Z-score smoothed over a 10 residue window
is plotted as function of the residue number. Low areas in the plot (below
-1.3) indicate "unusual" packing.
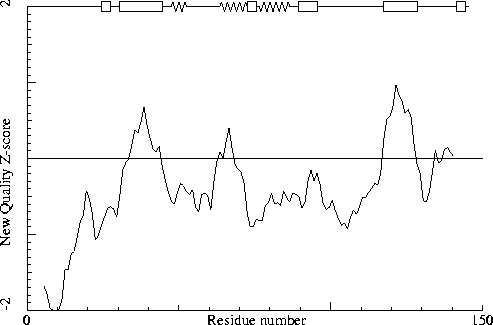
Chain without chain identifier
Warning: Backbone oxygen evaluation
The residues listed in the table below have an unusual backbone
oxygen position.
For each of the residues in the structure, a search was performed to find 5-residue stretches in the WHAT IF database with superposable C-alpha coordinates, and some constraints on the neighboring backbone oxygens.
In the following table the RMS distance between the backbone oxygen positions of these matching structures in the database and the position of the backbone oxygen atom in the current residue is given. If this number is larger than 1.5 a significant number of structures in the database show an alternative position for the backbone oxygen. If the number is larger than 2.0 most matching backbone fragments in the database have the peptide plane flipped. A manual check needs to be performed to assess whether the experimental data can support that alternative as well. The number in the last column is the number of database hits (maximum 80) used in the calculation. It is "normal" that some glycine residues show up in this list, but they are still worth checking!
108 VAL ( 385 ) 1.51 80
It is not necessarily an error if a few residues have rotamer values below 0.3, but careful inspection of all residues with these low values could be worth it.
139 GLU ( 417 ) 0.36
For this check, backbone conformations are compared with database structures using C-alpha superpositions with some restraints on the backbone oxygen positions.
A residue mentioned in the table can be part of a strange loop, or there might be something wrong with it or its directly surrounding residues. There are a few of these in every protein, but in any case it is worth looking at!
3 ARG ( 276 ) 0 88 THR ( 363 ) 0 115 MET ( 392 ) 0 116 CYS ( 393 ) 0 87 CYS ( 362 ) 2
Backbone conformation Z-score : 1.302
B-factor analysis
Note: Average B-factor OK
The average B-factor of buried atoms is within expected values for
a room-temperature X-ray study.
Average B-factor for buried atoms : 19.847
Note: Number of buried atoms with low B-factor is OK
For protein structures determined at room temperature, no more than
about 1 percent of the B factors of buried atoms is below 5.0.
Percentage of buried atoms with B less than 5 : 0.00
Note: B-factor distribution normal
The distribution of B-factors within residues is within expected
ranges. A value over 1.5 here would mean that the B-factors show
signs of over-refinement.
RMS Z-score : 1.468 over 992 bonds
Average difference in B over a bond : 1.94
RMS difference in B over a bond : 4.18
Note: B-factor plot
The average atomic B-factor per residue is plotted as function of
the residue number.
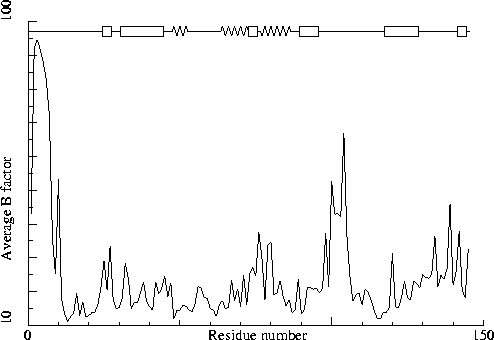
Chain without chain identifier
Hydrogen bond related checks
Error: HIS, ASN, GLN side chain flips
Listed here are Histidine, Asparagine or Glutamine residues for
which the orientation determined from hydrogen bonding analysis are
different from the assignment given in the input. Either they could
form energetically more favorable hydrogen bonds if the terminal
group was rotated by 180 degrees, or there is no assignment in the
input file (atom type 'A') but an assignment could be made. If a
residue is marked ``flexible'' the flipped conformation is only
slightly better than the non-flipped conformation.
1 HIS ( 274 ) 19 GLN ( 292 ) 28 ASN ( 301 ) 86 ASN ( 361 )
In the table below all normal histidine residues are listed. The assignment based on the geometry of the residue is listed first, together with the RMS Z-score for the fit to the Engh and Huber parameters. For all residues where the H-bond assignment is different, the assignment is listed in the last columns, together with its RMS Z-score to the Engh and Huber parameters.
As always, the RMS Z-scores should be close to 1.0 if the residues were restrained to the Engh and Huber parameters during refinement.
Please note that because the differences between the geometries of the different types are small it is possible that the geometric assignment given here does not correspond to the type used in refinement. This is especially true if the RMS Z-scores are much higher than 1.0.
If the two assignments differ, or the ``geometry'' RMS Z-score is high, it is advisable to verify the hydrogen bond assignment, check the HIS type used during the refinement and possibly adjust it.
1 HIS ( 274 ) HIS-E 0.68 12 HIS ( 285 ) HIS-E 0.72 26 HIS ( 299 ) HIS-E 0.70 49 HIS ( 324 ) HIS-D 0.75 HIS-E 0.76 60 HIS ( 335 ) HIS-E 0.78 109 HIS ( 386 ) HIS-E 0.80 HIS-D 1.04
Hydrogen bond donors that are buried inside the protein normally use all of their hydrogens to form hydrogen bonds within the protein. If there are any non hydrogen bonded buried hydrogen bond donors in the structure they will be listed here. In very good structures the number of listed atoms will tend to zero.
6 ARG ( 279 ) NH2 7 GLY ( 280 ) N 26 HIS ( 299 ) N 41 TYR ( 316 ) OH 59 ASN ( 334 ) N 64 LYS ( 339 ) N 77 ALA ( 352 ) N 89 ASP ( 364 ) N 91 GLN ( 366 ) N 92 ALA ( 367 ) N 116 CYS ( 393 ) N 120 ARG ( 397 ) N 130 GLN ( 407 ) N 142 ARG ( 420 ) N
The second part of the table mostly gives an impression of how well the model conforms to common refinement constraint values. The first part of the table shows a number of constraint-independent quality indicators.
Structure Z-scores, positive is better than average:
1st generation packing quality : -2.227 2nd generation packing quality : -3.147 (poor) Ramachandran plot appearance : -2.050 chi-1/chi-2 rotamer normality : -0.850 Backbone conformation : 1.302
Bond lengths : 0.765 Bond angles : 0.913 Omega angle restraints : 0.664 (tight) Side chain planarity : 0.078 (tight) Improper dihedral distribution : 0.577 B-factor distribution : 1.468 Inside/Outside distribution : 1.145