
In sequence alignments we need to cope with three problems:
- Trying out all alignments
- Scoring residue matches
- Scoring gaps
|
Multiple sequence alignments cannot be computed rigorously. Some approximations must be
made to get answers in an acceptable amount of time. These approximations sometimes
cause problems.
|
|
|
In sequence alignments we need to cope with three problems:
|
Problem one is solved by using a dynamic programming algorithm. Problem two is solved with a scorings matrix. Problem three is (too) difficult.
Align the following sequences:
>1 ASDFGRFGVCWPLKHTY >2 SDFGRFGVCWPLKHTY >3 ADFGRFGVCWPLKHTY >4 ASFGRFGVCWPLKHTY >5 ASDGRFGVCWPLKHTY >6 ASDFRFGVCWPLKHTY >7 ASDFGFGVCWPLKHTY |
You would expect the output to look like:
1 ASDFGRFGVCWPLKHTY 2 -SDFGRFGVCWPLKHTY 3 A-DFGRFGVCWPLKHTY 4 AS-FGRFGVCWPLKHTY 5 ASD-GRFGVCWPLKHTY 6 ASDF-RFGVCWPLKHTY 7 ASDFG-FGVCWPLKHTY
Unfortunately, Clustal "sorts" the sequences in the output. After unsorting, the output looks like (can be different depending on the Clustal server you choose, of course, I suggest you use the ClustalW2 server at the EBI):
1 ASDFGRFGVCWPLKHTY 2 -SDFGRFGVCWPLKHTY 3 -ADFGRFGVCWPLKHTY 4 -ASFGRFGVCWPLKHTY 5 ASDG-RFGVCWPLKHTY 6 ASDF-RFGVCWPLKHTY 7 ASDF-GFGVCWPLKHTY
Question 114: After aligning the seven sequences you see that two 'errors' are being made(these are not errors made by Clustal, but conceptual problems of the MSA problem):
And now even funnier. Lets randomize the sequences a bit:
>1 ASDFGRFGVCWPLKHTY >2 SDFGRFGVCWPLASDF >3 ADFGRFGVCWPLKRTH >4 ASFGRFGVCWPLPLMN >5 ASDGRFGVCWPLIHGV >6 ASDFRFGVCWPLARGM >7 ASDFGFGVCWPLPTHF |
Again, you see some "errors" around the gap, but this time even the order of alignments seems not to explain the problem. Conclusion: Alignments are more difficult than one thinks!
Question 115: Mention some reasons why multiple sequence alignment programs make errors.
Answer NOTE
|
If you have a set of sequences that includes a few that are far, far away in terms of evolutionary distance, you often can improve the alignment by removing those way-out sequences. |
The sequences required to run this exercise are available from the 'Exercise files' section under 'Miscellaneous' to the left of the screen.
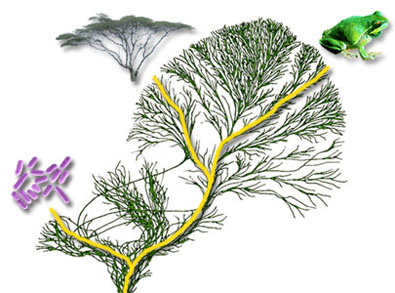
|
Figure 75. Sometimes you need a phylogenetric tree (dendogram) to get an impression about outlier sequences. In this exercise studying the alignment should suffice. |
Take the sequences of Set 1 and align them. Look at the alignment, it is a bewildering chaos. Why is this such a mess?
In Set 2 I have removed the sequences 1, 5, 11, and 18. (Why those 4?). Align the sequences in Set 2. This alignment looks much nicer already, doesnt it?
Look at the alignment of Set 2. It is much less of a mess already. But still, why am I going to remove the sequences 8, 19, 21, 23, and 25 to make Set 3?
Align Set 3. Look at the output. What is the lowest pairwise identity between sequences in set 3? The alignment contains only one dubious region. Find it and explain it.
Question 116: Explain what you did in this elaborate process of aligning and deleting sequences, why you did what you, and what is the result?
Answer NOTE
|
If you add information (in terms of more sequences) you can improve alignments. |
In this exercise we will each time add one sequence to the alignment. Run the alignments and cut-n-paste the results in a file (MS-Word, notepad, or whatever).
>S1 CWPAASTFLGAACPW >S2 CWPASSNGSACPW |
>S1 CWPAASTFLGAACPW >S2 CWPASSNGSACPW >S3 CWPRASTNAAACPW |
>S1 CWPAASTFLGAACPW >S2 CWPASSNGSACPW >S3 CWPRASTNAAACPW >S4 CWPRASSYNAAACPW |
>S1 CWPAASTFLGAACPW >S2 CWPASSNGSACPW >S3 CWPRASTNAAACPW >S4 CWPRASSYNAAACPW >S5 CWPASSTFNGAACPW |
Note the position of the gap and the position of the N-s in each consequetive alignment.
Question 117: Each time we added one sequence. I think the alignment also got better each time we added a sequence. Explain that.
Answer
EU name: EXE005
(Date: Aug 24 2016 EXE005A)
Question 118: In a previous exercise we improved the alignment by all the time deleting sequences, and in this exercise we improve by all the time adding sequences. So, what is going on? If I have a difficult alignment, should I add or delete sequences?
Answer