This is the sequence of a pseudo gene.
|
This is a manual analysis of some of the Alphafold2 models that got
a WHAT IF LR-score of ~100 (that is the worst LR-score that
WHAT IF can give). |
Sorry for the shitty graphics; but remember, you got these pages for free.
COMPND 2 MOLECULE: PUTATIVE PROTEIN T-ENOL; |
|
|
This is the sequence of a pseudo gene. |
COMPND 2 MOLECULE: G ANTIGEN 1; |

|
IUPRED2A predicts this protein to be natively unfolded over nearly the full length. |
COMPND 2 MOLECULE: CORNIFIN-A; |
|
SwissProt talks about repeats in the sequence. Normally, a repeat in a sequence means there either in no rigid 3D structure, or a structure with repeats. In case of the latter, no such structure exists in the PDB yet. Which means that homology modelling is not possible, not even if we would get a thousand times more sensitive BLAST. This should thus be modelled ab initio. |
COMPND 2 MOLECULE: MARCKS-RELATED PROTEIN; |
|
SwissProt tells us: Macrophage myristoylated alanine-rich C kinase substrate (Mac-MARCKS; MacMARCKS) that does something with Actin. And it has a dozen or so phosphoserines and phosphothreonines. |
COMPND 2 MOLECULE: G ANTIGEN 13; |

|
Looks like I totally accedentally picked a homolog of one of the examples above. Funny to see that it then also makes the same funny loop. |
COMPND 2 MOLECULE: PROTEIN GOLM2; |
|
A large part of this looks, according to BLAST, like 4TQL computationally designed three helix bundle (DE NOVO PROTEIN 11-JUN-14 4TQL ); three helix bundle albeit with only 21% sequence identity. So, classical modelling probably would have fared better on this one than Alpha fold. |
COMPND 2 MOLECULE: PROTEIN TNT; |

|
This is a DUF (Domain of Unknown Function) in PFAM. IUPRED2A predicts this protein to contain a significant fraction of natively unfolded stretches. There is vague structural similarity between the C-terminal half of this thing and some Myosin (for which no findable structure info exists). |
COMPND 2 MOLECULE: TRANSCRIPTION ELONGATION FACTOR A PROTEIN-LIKE 7; |
|
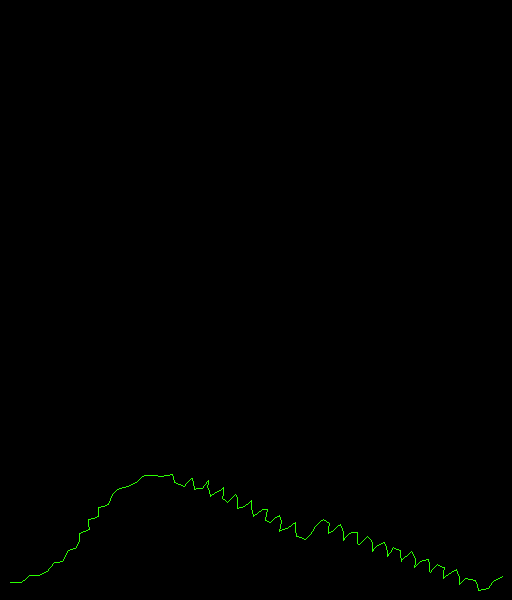
Some similarity is found by BLAST with 1WDZ (insulin receptor substrate p53). This is a thing with many big helices. It seems the secondary structure prediction capacity of Alphafold works well :-) |
|
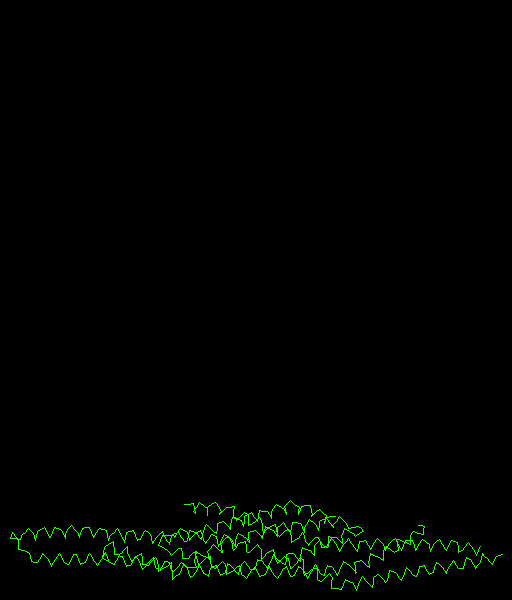
PDB file 1WDZ. |
COMPND 2 MOLECULE: COILED-COIL DOMAIN-CONTAINING PROTEIN 126; |

|
In SwissProt I also find: "sequence caution Sequence=AAQ96871.1; Type=Erroneous gene model prediction; Evidence={ECO:0000305};" |
COMPND 2 MOLECULE: KERATIN-ASSOCIATED PROTEIN 10-9; |

|
This is (part of) some keratin associated protein. It holds very many short repeats. The lower right corner looks a bit compact, but that is only because this is a 2D projection, there are no contacts there. |
COMPND 2 MOLECULE: PROBABLE FIBROSIN-1; |

|
Holds (according to Swissprot) several low complexity motifs and large disordered regions. The center looks a bit compact, but that is only because this is a 2D projection, there are no contacts there. |
COMPND 2 MOLECULE: G ANTIGEN 12B/C/D/E; |

|
Swissprot call the full protein disordered with as evidence MobiDB-lite. |