































# The angle between the carbonyls
oang = calcAngle(CO_PDB, CO_REDO)
# The angle between the amides
hang = calcAngle(NH_PDB, NH_REDO)
# Omega difference
opdb = calcTorsion(CACNCA_PDB)
oredo = calcTorsion(CACNCA_REDO)
odif = abs(opdb - oredo)
# C-alpha displacement
cadif_i-1 = distance(CA_i-1_PDB, CA_i-1_REDO)
cadif_i = distance(CA_i_PDB, CA_i_REDO)
cadif = max(cadif_i-1, cadif_i)
if oang > 120 or hang > 120 or odif > 120:
if cadif > 1.0:
appendFlipType('displaced_')
endif
if odif > 120:
# cis-trans flip
if abs(oredo) > abs(opdb):
appendFlipType('ct')
else:
appendFlipType('tc')
endif
# CO-flip, NH-flip, or hard to determine automatically?
if oang > 120 and hang < 60:
appendFlipType('+')
else if hang > 120 and oang < 60:
appendFlipType('-')
else
appendFlipType('_irregular')
endif
else if odif < 60:
# peptide plane flip
if oang > 120 and hang > 120:
appendFlipType('tt+')
else
appendFlipType('tt+_irregular')
endif
else
appendFlipType('omega_irregular')
endif
endif
Introduction
Peptide bonds can have two conformations. The torsion angle ω (Cαi-1-Ci-1-Ni-Cαi) can be around 0°, cis, or around 180°, trans. The peptides in the protein structures in the Protein Data Bank (PDB) almost exclusively contain the trans conformation. However, some of those conformations are incorrect and should actually be cis. Other trans peptide planes should stay trans but need to be rotated ~180° about the Cα-Cα axis. This website details the methodology used to the predict cis↔trans flips and peptide plane flips in the backbone of protein structures, and contains information supplementary to the manuscript 'Detection of trans–cis flips and peptide plane flips in protein structures' by Wouter G. Touw, Robbie P. Joosten and Gert Vriend. The method predicts ~70K peptide plane flips and ~5K trans → cis flips.
Predict flips
There are several options to predict flips:
WHAT_CHECK
The FLPCHK validation routine of WHAT IF's CHECK menu.
Web service
The ShowPepFlips WHAT IF Web Services (WIWS) option allows SOAP and REST requests.
Web server
The flip checks are on the WHAT IF web servers
cis ↔ trans and peptide plane flips
We have adopted a systematic naming system for flips and non-flips. The flip type is indicated by three characters. The first character indicates the starting omega conformation (either t for trans or c for cis). The second character indicates the correct omega conformation (again t or c). The third character indicates whether the carbonyl 'flips' (+) or not (-). For cis-trans flips the third character implies the reverse for the N-H, i.e. tc- involves a NH-flip. Both an NH-flip and CO-flip occur when peptide planes are flipped (tt+). Theoretically there are 6 possible flip types and tt- and cc- designate correct trans and cis peptides:
| Flip type | Explanation |
|---|---|
| tt- | The peptide conformation is correct and should stay trans |
| tt+ | A peptide plane flip (~180° crankshaft motion about Cα-Cα axis); both CO-flip and NH-flip. |
| tc- | trans to cis with NH-flip |
| tc+ | trans to cis with CO-flip |
| cc- | The peptide conformation is correct and should stay cis |
| cc+ | The entire Cα-C-N-Cα unit theoretically would rotate ~180° |
| ct- | cis to trans with NH-flip |
| ct+ | cis to trans with CO-flip |
tt+
tc-
tc+
ct-
ct+
Prediction
Training data
Pairs of X-ray structures solved at 3.5 Å or better, containing at least 25 amino acids in at least one chain, for which a DSSP file (Kabsch & Sander, 1981) exists, and that contained at least one trans - cis or peptide plane flip between PDB and PDB_REDO were obtained from the releases of 20-10-2014. From these PDB files stretches of four canonical residues were selected that had all atoms present with non-zero B-factor and full occupancy; no covalently bound atoms were allowed other than the continuation of the chain; all torsion angles and the DSSP secondary structure must be determinable; the four amino acids were neither N- or C-terminal, nor adjacent to a chain break. A training set was obtained by comparing peptide conformations in the pairs of PDB and PDB_REDO structures. The procedure calculates three values 1) ΔC=O, which is the angle between the PDB carbonyl and the PDB_REDO carbonyl after optimal superposition; 2) ΔN-H, which is the angle between the N-H pair; 3) Δω which is the ω torsion angle difference . If Δω is big, a cis-trans NH-flip (tc- or ct-) is assigned when ΔN-H is big and ΔC=O is small, and a CO-flip (tc+ or tc-) is assigned when ΔC=O is big and ΔN-H is small. If Δω is small and both ΔN-H and ΔC=O are big a peptide plane flip is assigned. It was found that the best assignments were obtained when ‘big’ was defined as being greater than 120° and ‘small’ was defined as being less than 60°. Irregular cases were excluded from the training examples. For irregular cases a) ΔN-H or ΔC=O or Δω is big but other criteria are not met; b) either one of the Cα atoms flanking the peptide plane has been superposed with more than 1 Å displacement. Click on the bar to show/hide the pseudo-code for determining the flip types.
Obviously, the different flip classes and the correct classes (tt- and cc-) are not equally distributed since the overwhelming majority of the PDB peptides have the correct conformation. It is well known that it is notoriously hard for machine learning algorithms to deal with highly skewed data, for example because always predicting the majority class still gives almost perfect prediction accuracy. This is also known as the imbalance problem. Popular strategies include under-sampling of the majority class, over-sampling of the minority class, a combination of both. We found that randomly downsampling the majority class to the size of the minority class to obtain a balanced training set worked well for training Random Forests (see below). When repeated with different random seeds, very similar results were obtained. Training with unbalanced data using estimated class priors did not work well in our hands.
Download training dataTest data
The test cases were manually validated and re-refined when necessary. Note that some of the validated peptides do not conform to all of the training set criteria. For example, some residues in the tetra peptides may be incomplete, are bound to something, etc. When determining the performance in the validation process, these cases have not been included.
Download test dataFeature changes
An incorrectly modeled peptide usually causes problems for the local backbone. The peptide has to be accomodated, also if little space is available, causing a fight between the X-ray data and refinement restraints. This causes strain that shows up in several features describing the local backbone conformation. If the peptide conformation is corrected by a cis↔trans flip or a peptide plane flip and the structure is re-refined, the strain will be relieved and the backbone parameters will adopt their normal values. The figures below show the change in continuous features (described in the Features section) upon flipping and re-refinement by PDB_REDO for x-X-Xnpg-x (X: any residue; Xnpg: any residue except Pro and Gly) tt-/tt+/tc-/tc+, x-X-Pro-x tt-/tc-/tc+, and x-X-Gly-x tt-/tt+ peptides in the test data. The lines are Gaussian kernel density estimates for the number of cases indicated in the legend for each flip class. The feature distributions in PDB structures are indicated with solid lines. The PDB_REDO distributions are indicated with dotted lines. For example, in the ψi (psi) plot for X-Xnpg the PDB_REDO distributions have been shifted for the 56 tt+, 49 tc- and 10 tc+ cases with respect to the PDB ψi distribution. The 'difference' plots show the feature distributions for PDB_REDO - PDB (i.e. 0 means no difference before and after correction). Click on the bar to show/hide the figures.
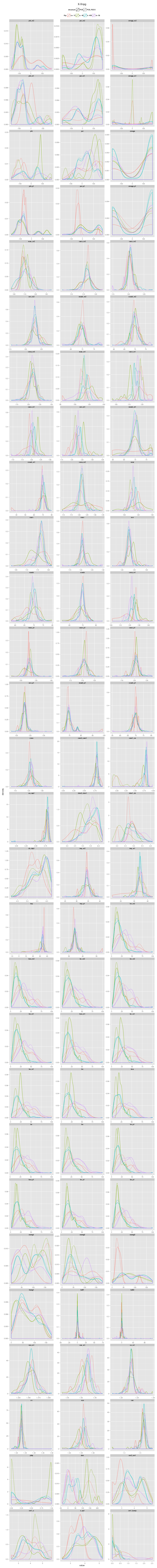
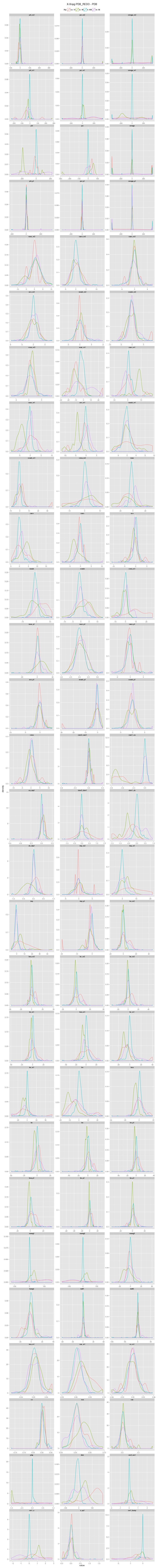
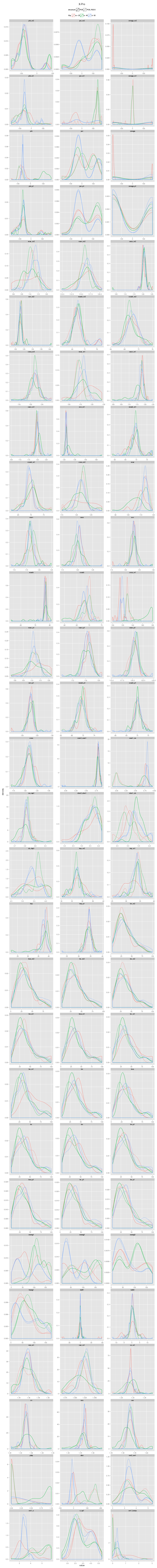
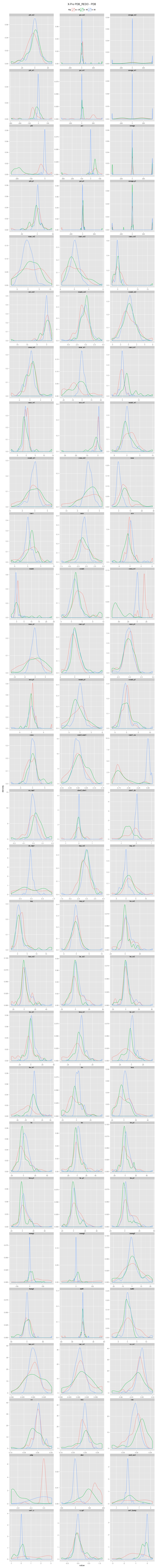
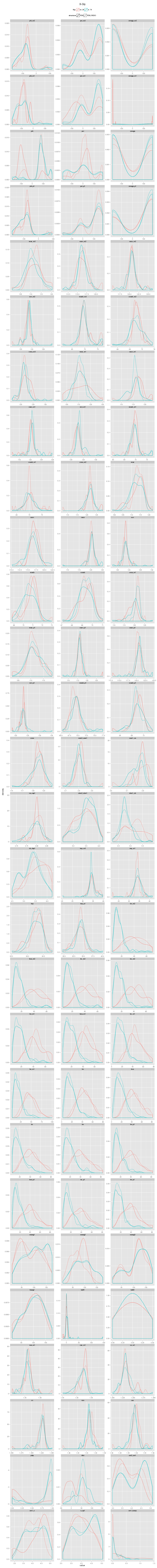
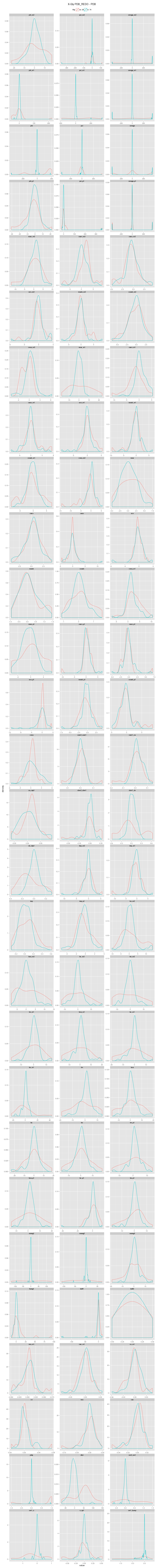
Features
The features that can capture the difference between an incorrect and correct peptide bond conformation belong to several feature groups and have been calculated for four amino acids surrounding the peptide bond. The feature groups are angles, torsion angles, distances, chiral volumes, B-factors, secondary structure, and a few other groups explained in the table. Rather than B-factors from PDB files, we used B-factors from BDB files. The BDB is a databank that contains PDB files with consistent B-factors (Touw & Vriend, 2014). Most features were calculated by WHAT IF. Click on the bar to show/hide the comprehensive list of all features and their explanation. Note that the definition of ω for residue i in WHAT IF is equal to the standard definition of ω for residue i+1.
Training
4 different training data sets were constructed: X-Xnpg tc-, X-Xnpg tt+, X-Pro tc+ and X-Gly tt+. For all these training sets the negative class is tt-. For all 4 training sets a Random forest (RF; Breiman, 2001) classifier was constructed. The flip type-specific classifiers were later combined into one classifier per residue class. This strategy optimally made use of the available training examples (e.g. many more tt- and tt+ cases could be used for X-Xnpg flips than could have been used with a multi-class training with a balanced data set). A description of RF is available here. In short, an RF is an ensemble of decision trees. Each classification tree is constructed using a random subset of training examples and a random subset of features. The individual trees are so-called weak learners; the correlation between them is relativey small. Every tree predicts the class of a training example. The ensemble individual trees, the RF, classifies new cases by collecting votes from each tree. A threshold determines the fraction of votes that is needed to predict the class. The combination of enough sufficiently uncorrelated weak learners increases the classification strength of the ensemble and usually leads to a robust classifier. The RF models were trained using the randomForest and caret R packages. The RF parameter mtry was tuned using 5-fold cross-validation that was repeated 20 times with different random seeds. The classification performance on the training examples was measured by and/or optimized against the cutoff-independent area under the ROC curve , against the area under the precision-recall curve, and/or against the Matthews correlation coefficient and euclidian distance to perfect specificity and sensitivity at the optimal threshold determined by tuning across 40 different threshold values between 0.5 and 1 (code examples here and here).
The final classifiers were constructed using all training data with parameters from the best-performing cross-validation models. The resampling performance for the final classifiers is shown in the table below (click on the bar to show/hide the table). The AUC under the ROC curve has been calculated by the ROCR package. The rocplus package was used to obtain the AUC under the precision-recall curve.
Features important for correct classification of training examples are shown below. These figures show either the overall mean and sd permutation importance or scaled class-specific importance.
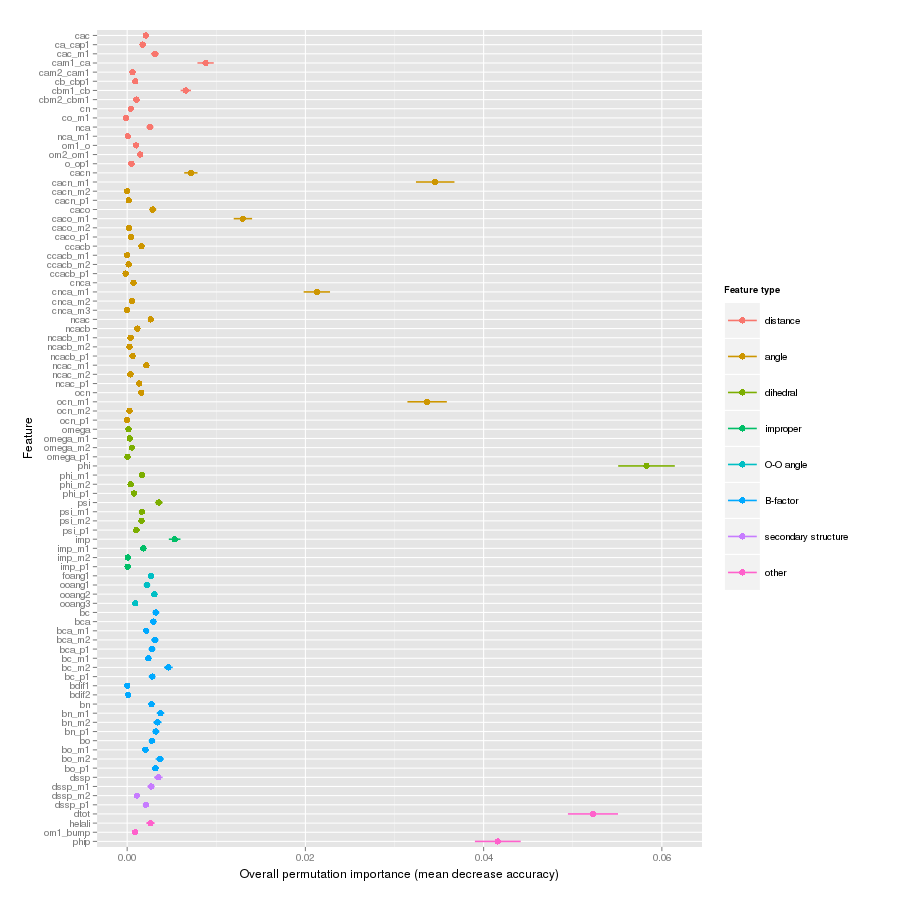
X-Xnpg tc-
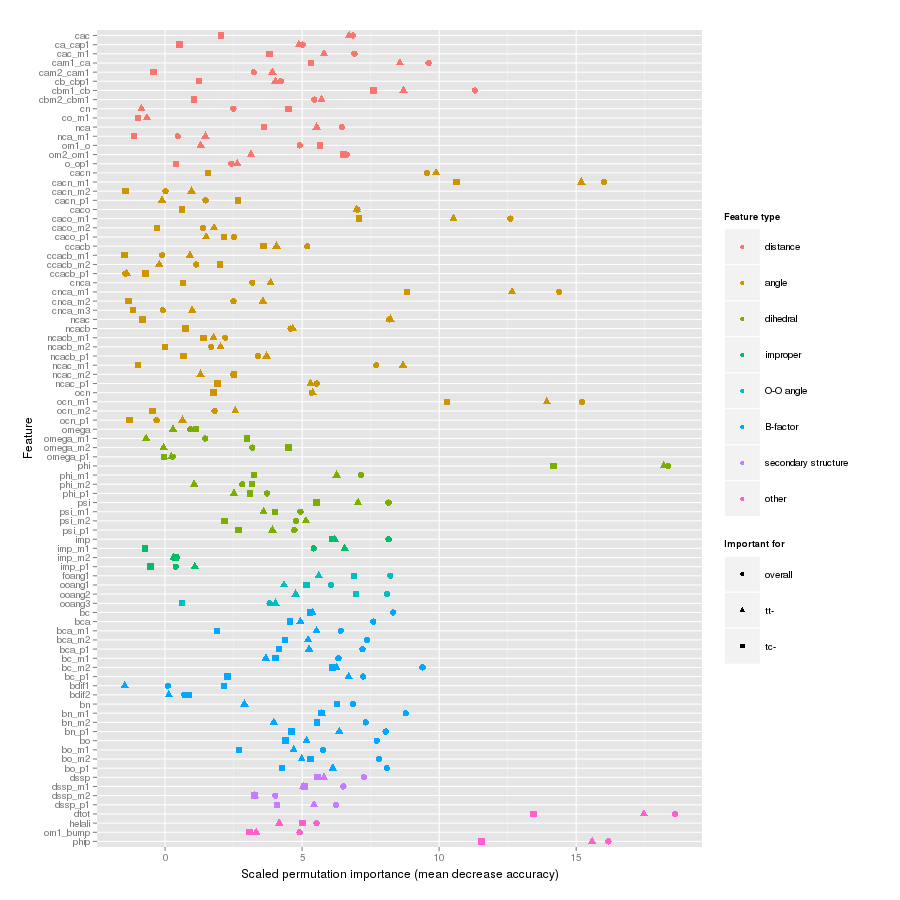
X-Xnpg tc-
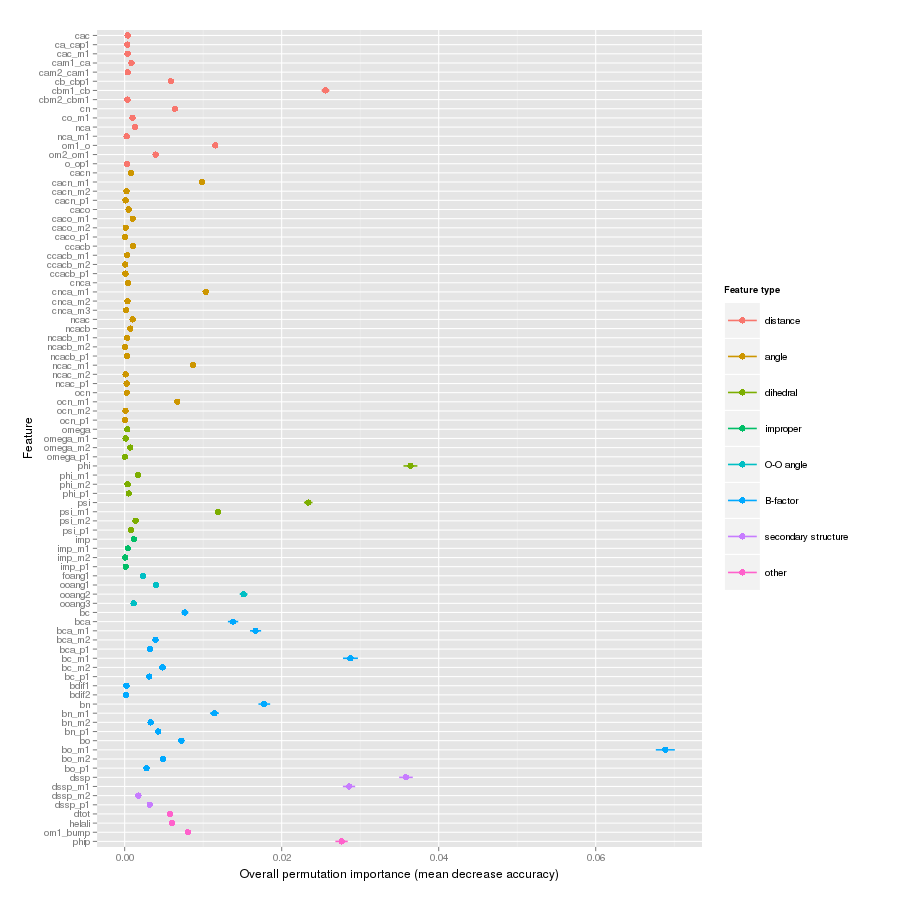
X-Xnpg tt+
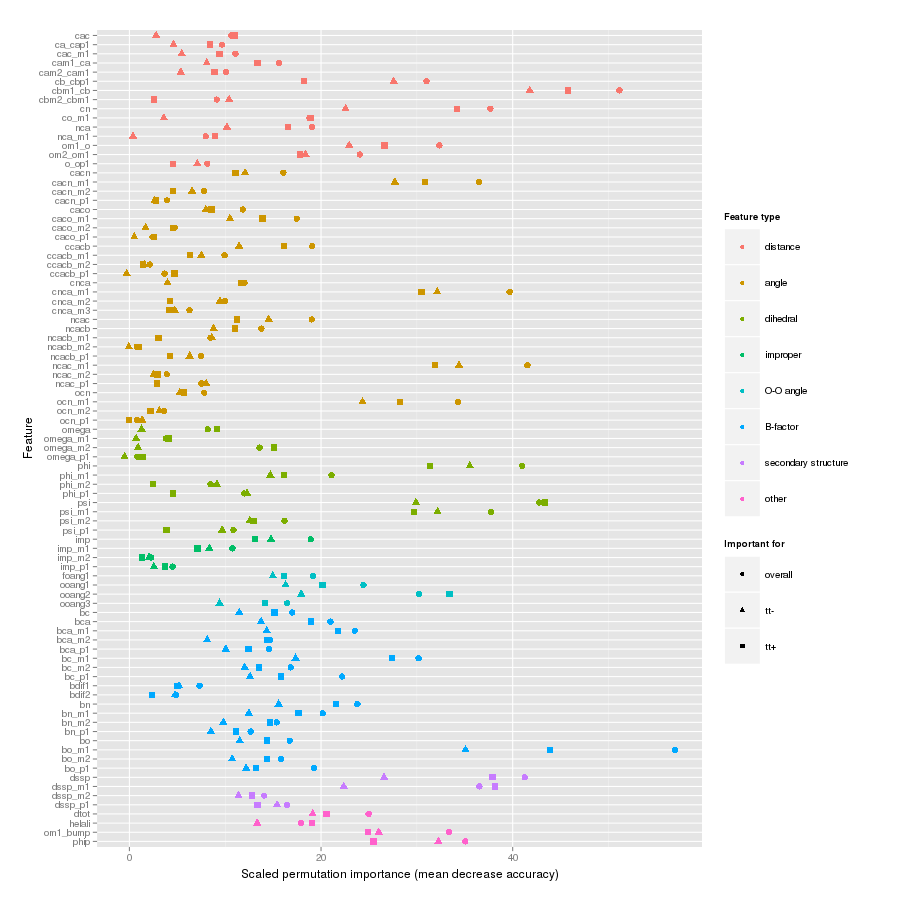
X-Xnpg tt+
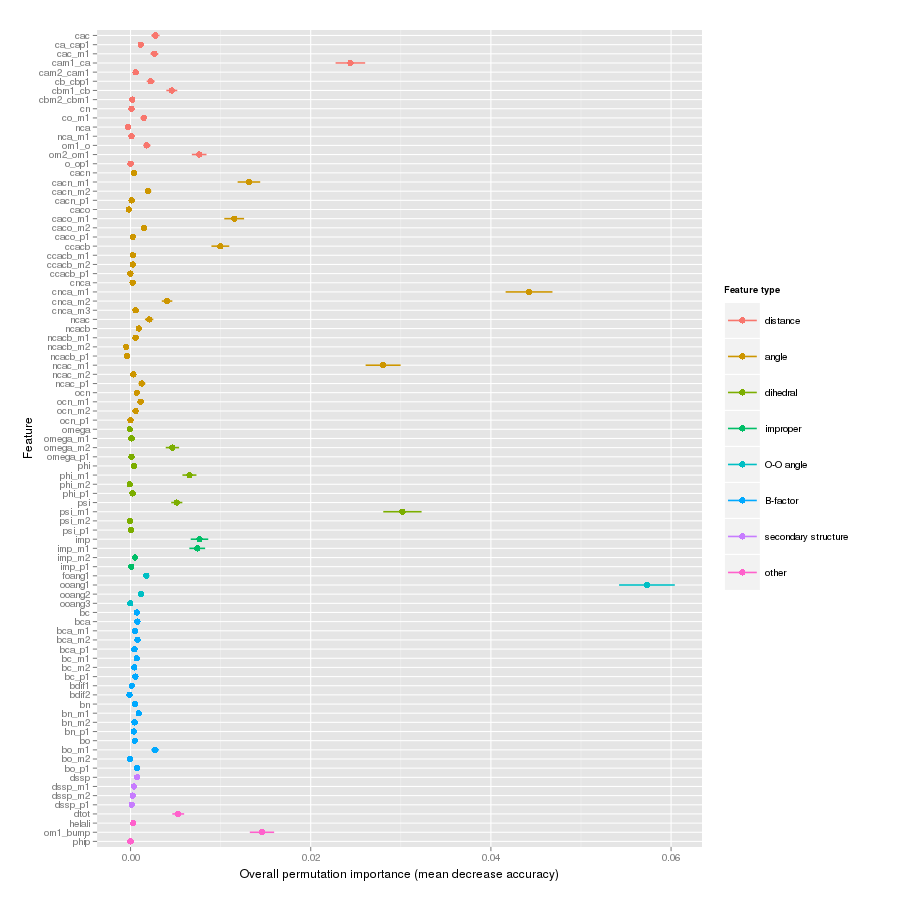
X-Pro tc+
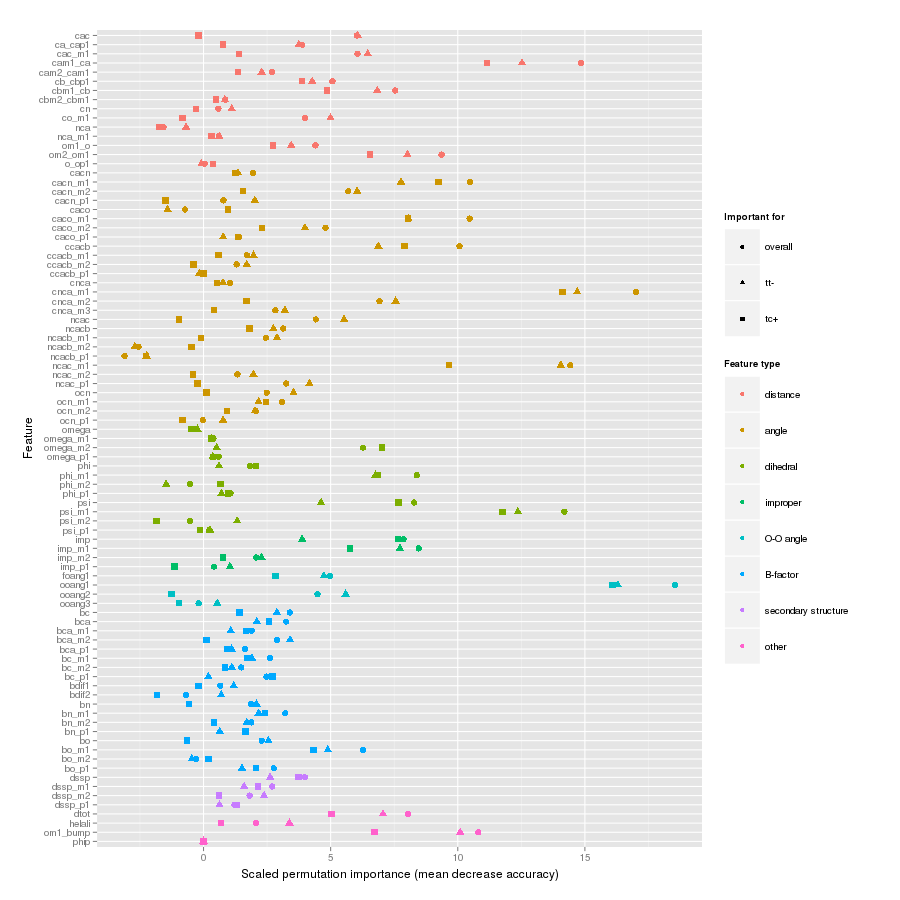
X-Pro tc+
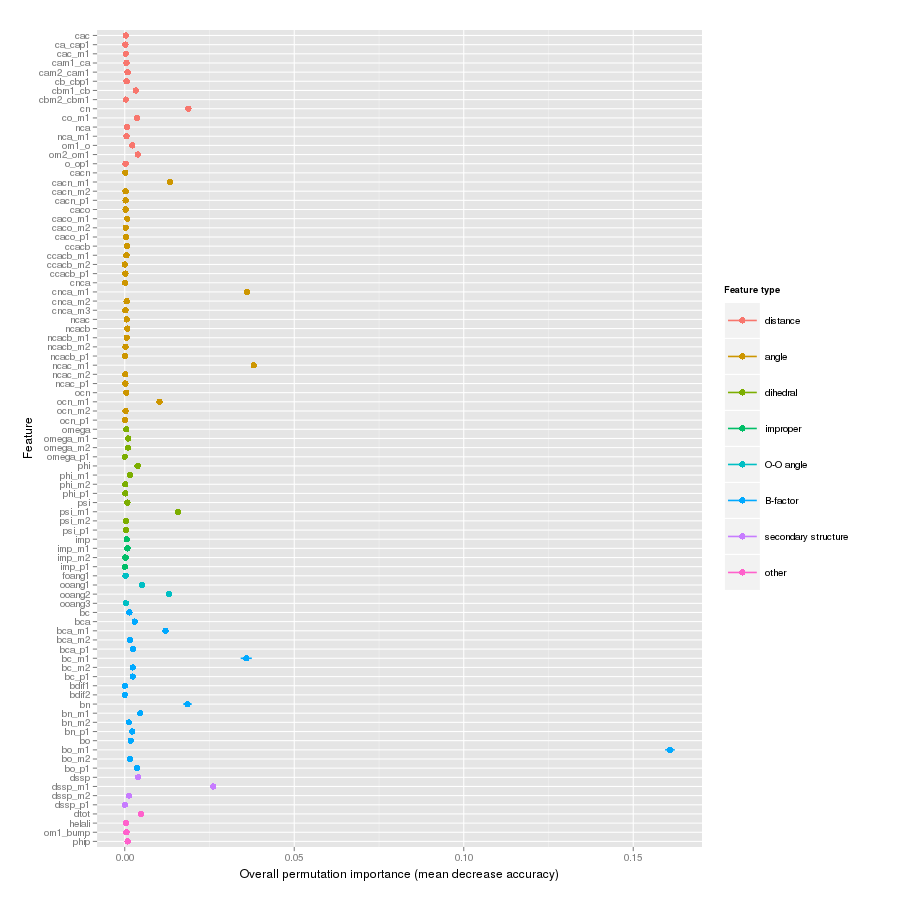
X-Gly tt+
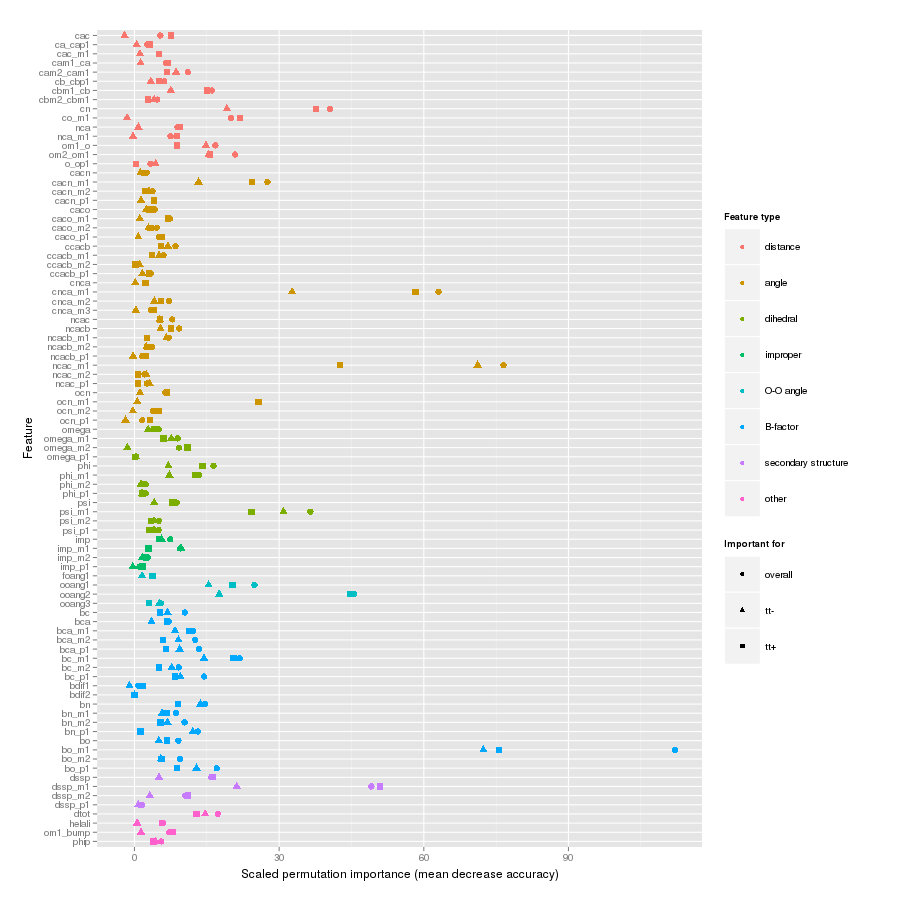
X-Gly tt+
The classifiers have been converted (automatically) to FORTRAN IF/ELSE statements for inclusion in WHAT_CHECK:
X-Pro prediction
All X-Pro tc- cases in the test set derived from the PDB-PDB_REDO comparision had a positive φi angle and could be separated perfectly from tt- and tc+ cases, for which the average φ is always around -60°. The rule φ > 0° misclassifies two tc+ instances . When this rule was applied to the entire PDB we also found examples of X-Pro with positive φ angles other than tc- cases. Incorrect chirality of the nitrogen atoms resulted for example from strain in the local backbone because residues i+1 or i-1 needed to be flipped. The class of trans X-Pro residues with N-chirality problems was called nch. The average φ is 96° for tc- and 12° for nch. The nch cases could be separated from tc- cases by a simple rule: if the angle N-Cα-C is large (> 112.47°) and the bump score of the oxygen in the peptide plane is large (> 0.26 WHAT IF bump score units), then the X-Pro with a positive φ is not a cis peptide in need of a tc- flip but a trans-Pro with N-chirality problems. This rule was found by visual inspection of the data using RFScout, a tool that allows the creation of Simple Decision Models. The WHAT_CHECK code for this classifier has been hand-written and not generated automatically.Cis → trans
We detected only 44 cis → trans flips in the entire PDB. This means not enough data was available to create accurate classifiers. Our method therefore does not include cis → trans flip prediction. Nevertheless we observed that the Cαi-1-Cαi distance and the Ci-1-Ni-Cαi angle tend to be larger for ct+ and ct- cases than for normal cc- tetramers. This can be expected because the data works against the cis restraints. For X-Pro we also observed that the angle τ (Ni-C&alphai-Ci) may help to separate ct+, ct-, and cc- cases:
Validation
The five remaining classifiers were tested against an independent test set. The performance of both individual and combined prediction models are shown in the tables below. These tables also show the performance on the subset of test cases that does not include NCS-related flip examples (only the first example in a structure is retained). We deliberately included a few NCS-related examples to observe how sensitive the RF are to small inter-chain variation.
| X-Xnpg tt+ | X-Xnpg tc- | X-Xnpg WH’ | X-Xnpg WH | X-Pro tc+ | X-Gly tt+ | |
|---|---|---|---|---|---|---|
| ROC AUC | 0.995/0.994 | 0.983/0.976 | 0.966/0.979 | 0.966 | 0.941/0.938 | 0.982/0.977 |
| Precision-recall AUC | 0.995/0.994 | 0.983/0.976 | 0.966/0.979 | 0.966 | 0.941/0.938 | 0.982/0.977 |
| Matthews correlation coefficient | 0.911/0.897 | 0.892/0.829 | 0.822/0.779 | 0.315 | 0.852/0.844 | 0.928/0.907 |
| Accuracy | 0.979/0.978 | 0.963/0.968 | 0.935/0.954 | 0.803 | 0.924/0.926 | 0.971/0.965 |
| True positive rate (sensitivity/recall) | 0.952/0.938 | 0.884/0.837 | 0.909/0.884 | 0.124 | 0.824/0.795 | 0.964/0.952 |
| False positive rate (fallout) | 0.012/0.016 | 0.014/0.016 | 0.057/0.038 | 0.0 | 0.0/0.0 | 0.026/0.031 |
| True negative rate (specificity) | 0.983/0.984 | 0.986/0.984 | 0.943/0.962 | 1.0 | 1.0/1.0 | 0.974/0.969 |
| False negative rate (miss rate) | 0.048/0.062 | 0.116/0.163 | 0.091/0.116 | 0.876 | 0.176/0.205 | 0.036/0.048 |
| Positive predictive value (precision) | 0.894/0.882 | 0.947/0.857 | 0.821/0.731 | 1.0 | 1.0/1.0 | 0.931/0.909 |
| Negative predictive value | 0.993/0.992 | 0.967/0.981 | 0.973/0.986 | 0.798 | 0.881.0.895 | 0.987/0.984 |
| Actual class | ||||
|---|---|---|---|---|
| Predicted class | tt- | tt+ | tc- | tc+ |
| tt- | 405 | 3 | 12 | 6 |
| tt+ | 7 | 59 | 2 | 5 |
| tc- | 6 | 0 | 107 | 1 |
| tc+ | 0 | 0 | 0 | 0 |
| Actual class | |||||
|---|---|---|---|---|---|
| Predicted class | tt- | tt+ | tc- | tc+ | nch |
| tt- | 89 | 0 | 0 | 12 | 0 |
| tt+ | 0 | 0 | 0 | 0 | 0 |
| tc- | 0 | 0 | 58 | 0 | 1 |
| tc+ | 0 | 0 | 0 | 54 | 0 |
| nch | 0 | 1 | 0 | 2 | 22 |
Usage
Flips can be predicted with the FLPCHK validation routines in WHAT_CHECK, the ShowPepFlips WHAT IF web services option, or the WHAT IF web server. The response of our RF classifiers was translated to qualitative measures of severity based on the validation set. All available methods therefore also report if it is ‘highly unlikely’, ‘unlikely’, ‘somewhat likely’, ‘likely’, or ‘highly likely’ that a peptide needs to be flipped or requires the attention of a structural biologist. The 'highly likely' category has no FP in the test set, the 'likely' category has between 0 and 2% FP in the test set, the 'somewhat likely' category has up to 10% FP, the 'unlikely' category has beteen 0 and 2% FN in the test set, and the 'highly unlikely' category has 0 FN in the test set.Simulation
In an effort to classify the small classes (and possibly address the imbalance problem further), X-Xnpg tc+, ct-, and ct+ errors were simulated. The errors were simulated for well-defined peptides selected from medium-sized single chain structures solved at a resolution better than 2.0 Å. WHAT IF (Vriend, 1990) flipped the peptides and relaxed the strain in a 20-residue window spanning the local backbone of the peptides. Subsequently, the flipped peptides were refined. After refinement only a small fraction of the peptide conformations still had a wrong conformation. Classifiers were constructed with 114 fabricated tc+ cases, but a two-class classifier could only correctly classify a third of the true tc+ cases, and none of the true tc+ cases could be distinguished by the four-class classifier. Furthermore, the classifiers did not pick up any previously unrecognized tc+ in the PDB. Finally, the simulated tc+ cases had a broad distribution and showed overlap with all flip classes when they were projected in the Principal Component Analysis space of validated tt-, tt+, tc-, and tc+ cases. Although the number of true tc+ cases very low, the simulated tc+ cases seemed to be more similar to tc- and tt- cases than to tc+ cases in most dimensions. In summary, the simulated flips were not representative of actual flips and could therefore not be used to train classifiers.
Re-building and re-refinement: changes in reciprocal- and real-space coefficients
Single peptide and cis-trans flips will probably have a small effect on the R-factors. The local changes in the protein backbone are expected to lead to an increase in local real-space correlation coefficient. We here present the flip correction and re-refinement of 1hi8, 1i6n, 2z81 and 1pe9 as examples.1hi8
The RNA dependent RNA polymerase from dsRNA bacteriophage φ6 PDB structure 1hi8 has been solved at 2.50 Å. The reported R-/R-free factors are 0.280/0.316.Two cis-peptides are reported in the PDB file:
CISPEP 1 ILE A 96 PRO A 97 0 0.03
CISPEP 2 ILE B 96 PRO B 97 0 0.04
both Ile-Pro peptide planes fit the electron density well:


However, 8 tt+ and Pro tc+ flips are necessary in both chain A and B (a total of 16 flips):
GLY 92 - A tt+
GLY 92 - B tt+
ASP 137 - A tt+
ASP 137 - B tt+
PRO 154 - A tc+
PRO 154 - B tc+
ALA 210 - A tt+
ALA 210 - B tt+
LEU 391 - A tt+
LEU 391 - B tt+
MSE 406 - A tt+
MSE 406 - B tt+
ARG 584 - A tt+
ARG 584 - B tt+
LYS 627 - A tt+
LYS 627 - B tt+
These screenshots show for either the A or the B chain their PDB conformation.




The WHAT_CHECK validation report flags Asp 137 and Pro 154 for having unusual C-N-Cα bond angles and Leu 391 for having unusual torsion angles. Furthermore, Gly 92, Als 210 and Leu 391 have unusual φ/ψ combinations. The buried hydrogen bond donor Gly 92 N (see figure above) is picked up, as well.
10 cycles TLS refinement and 50 cycles restrained refinement in REFMAC with these parameters results in the following re-refinement statistics:
| Structure | R-work reported | R-free reported | R-work initial | R-free initial | R-work final | R-free final | Work CC initial | Free CC initial | Work CC final | Free CC final | Work CC Z-score | Free CC Z-score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1hi8 | 0.280 | 0.316 | 0.2760 | 0.3082 | 0.2579 | 0.2868 | 0.8893 | 0.8636 | 0.9025 | 0.8836 | 11.84 | 3.46 |
| 1hi8 rebuilt | N/A | N/A | 0.2749 | 0.3044 | 0.2556 | 0.2825 | 0.8903 | 0.8660 | 0.9043 | 0.8864 | 12.72 | 0.07 |
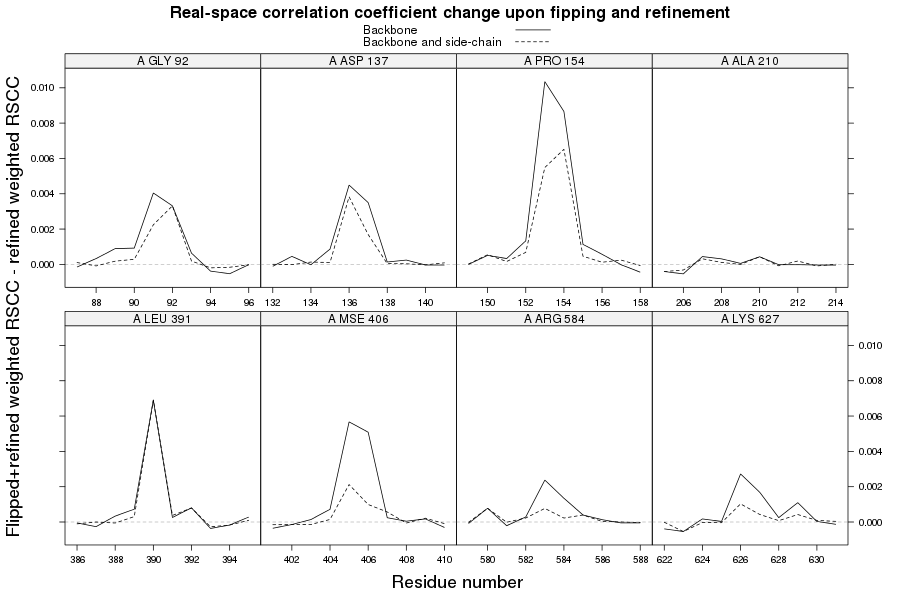
The corrections lead to positive RSCC difference around most flipped peptide bonds, which indicates that the local fit is improved. The improvement is dominated by the backbone atom increase in RSCC. As expected, the fit around Ala 210 is not better in the rebuilt/re-refined structure than in the re-refined structure because the conformation is correct in both structures.
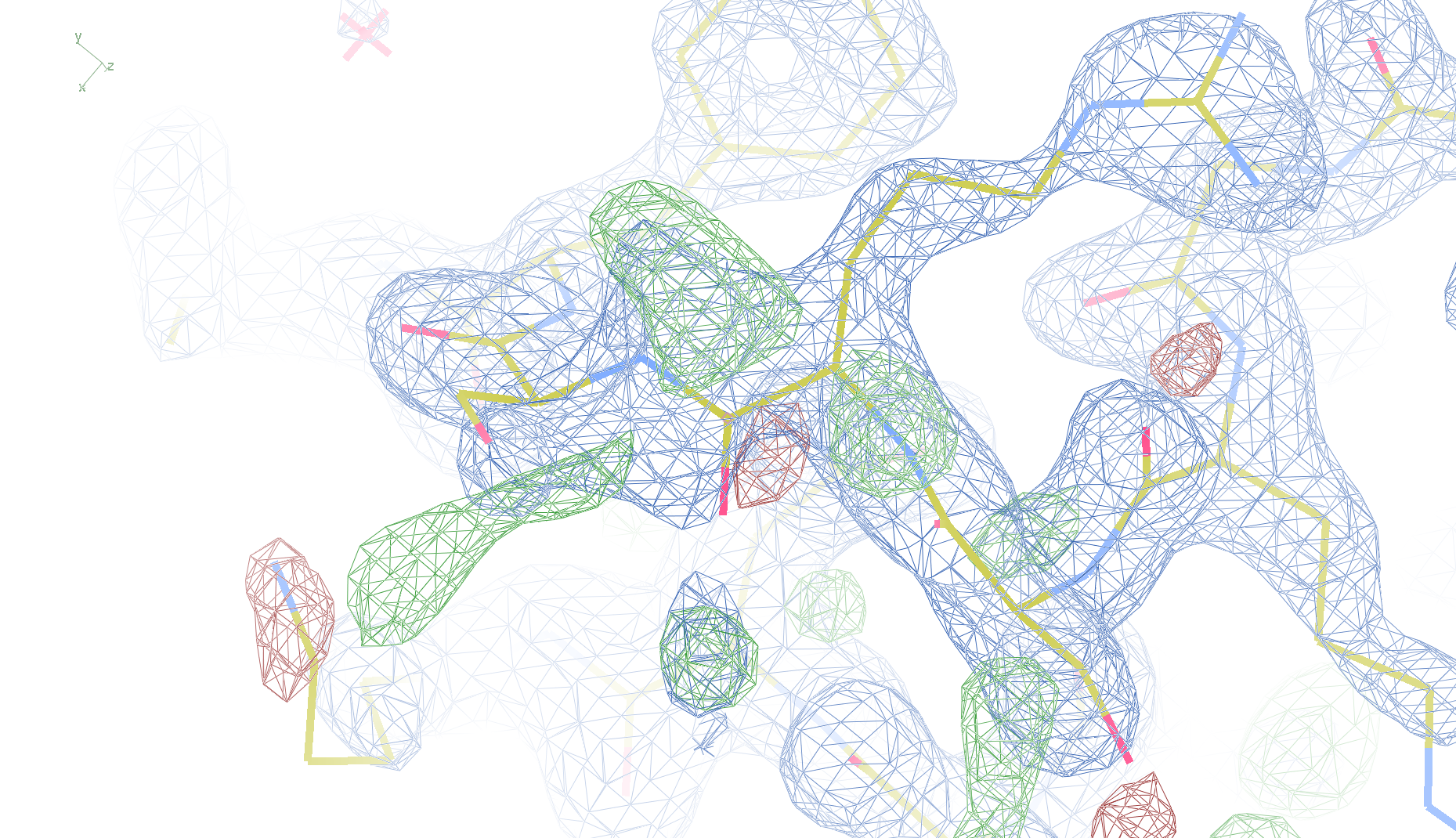
1i6n
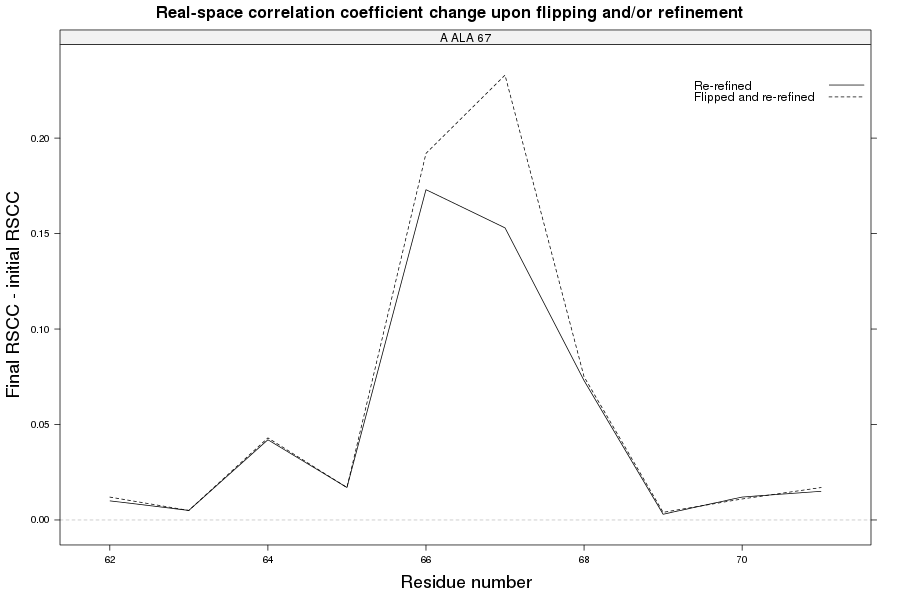
The crystal structure of Bacillus subtilis ioli 1i6n has been solved at 1.80 Å. The reported R-/R-free factors are 0.201/0.238. CISPEP records are absent in the PDB file.The following trans → cis NH-flip will correct many local problems:
1i6n ALA 67 - A tc-
The local backbone around Ala 67 is also wrong in 1i60 (see above), the
model that was used to solve 1i6n by molecular replacement. The
difference density shows many peaks around Asn 66, Ala 67 and Leu68
from the PDB model (first picture).




| Structure | R-work reported | R-free reported | R-work initial | R-free initial | R-work final | R-free final | Work CC initial | Free CC initial | Work CC final | Free CC final | Work CC Z-score | Free CC Z-score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1i6n | 0.201 | 0.238 | 0.1946 | 0.2280 | 0.1762 | 0.2089 | 0.9294 | 0.9039 | 0.9394 | 0.9170 | 10.19 | 2.26 |
| 1i6n rebuilt | N/A | N/A | 0.1929 | 0.2257 | 0.1761 | 0.2089 | 0.9305 | 0.9051 | 0.9393 | 0.9169 | 9.03 | 2.05 |
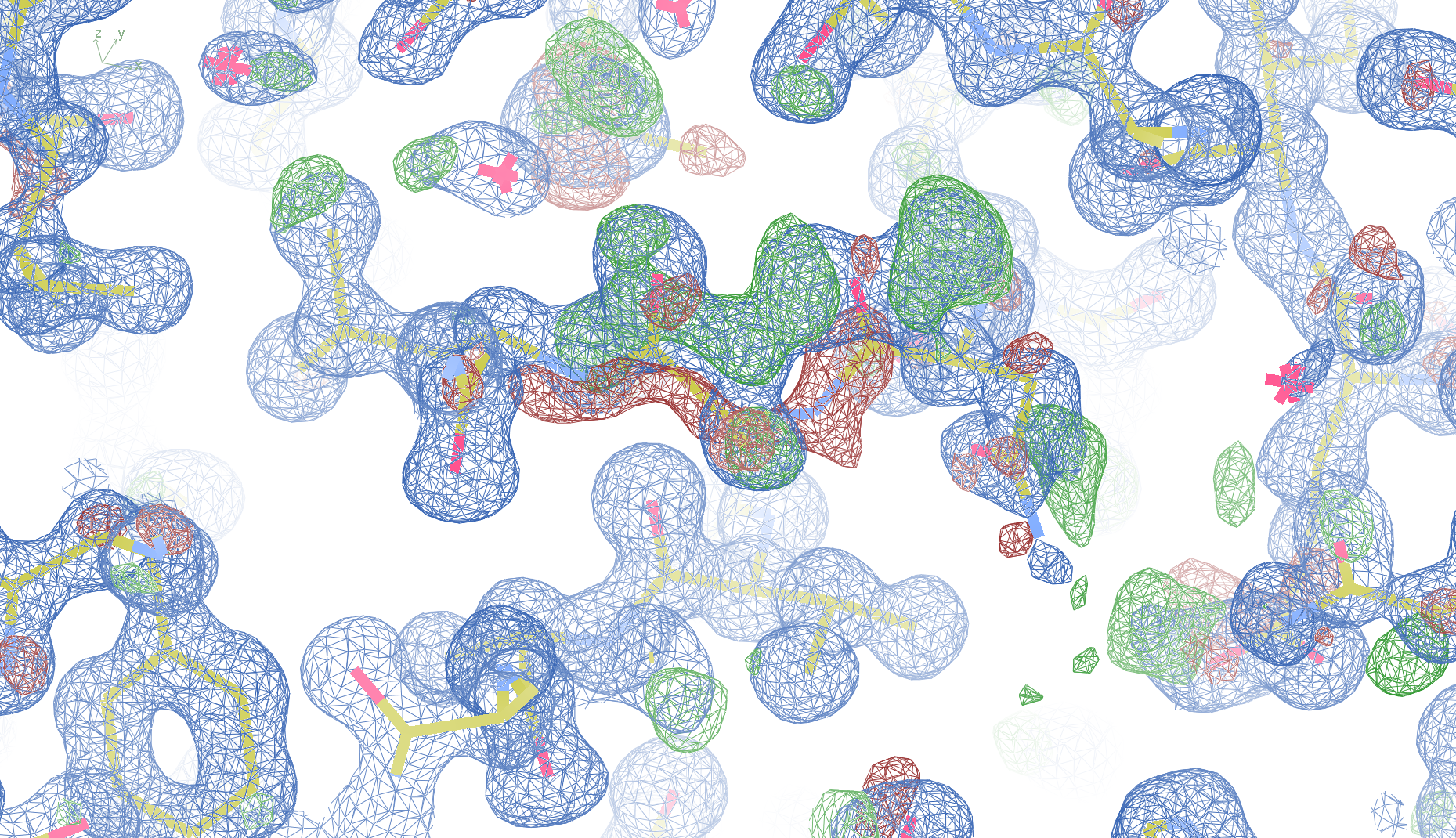
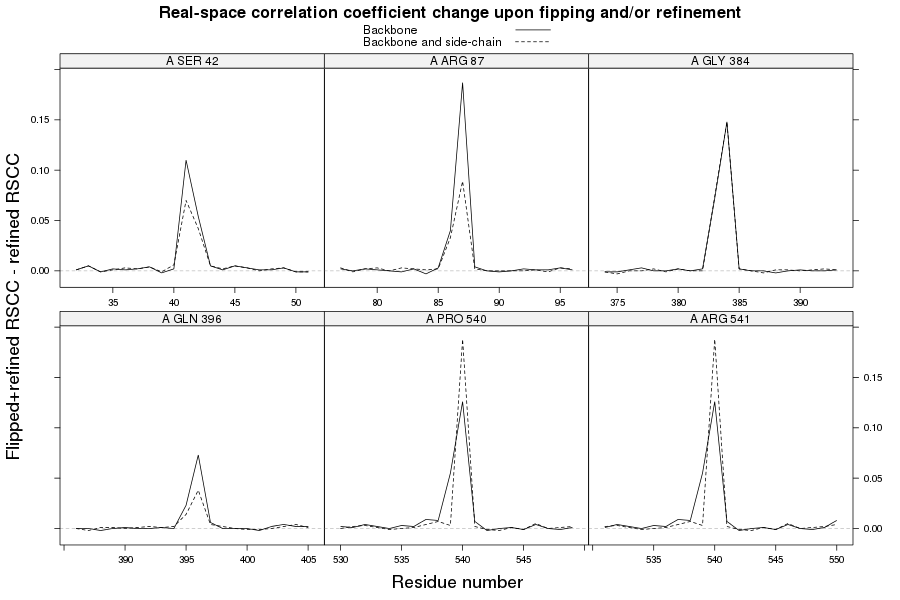
2z81
The crystal structure of the Toll-like receptor 2 2z81 has been solved at 1.80 Å and refined to reported R-/R-free factors of 0.213/0.232. CISPEP records are absent in the PDB file.1 Pro tc+ and 5 tt+ flips are necessary in 2z81:
2z81 SER 42 - A tt+
2z81 ARG 87 - A tt+
2z81 GLY 384 - A tt+
2z81 GLN 396 - A tt+
2z81 PRO 540 - A tc+
2z81 ARG 541 - A tt+
These screenshots show their PDB conformation.

The WHAT_CHECK validation report reports τ (N-Cα-C) angle problems around these bonds as well as poor φ/ψ combinations. Arg 87, Gly 384 and Gln 396 are listed as buried unsatisfied hydrogen bond donors.
The re-refinement statistics after 10 cycles of TLS refinement and 50 cycles restrained refinement in REFMAC with these parameters are slightly better for the rebuilt/re-refined structure than the re-refined-only structure:
| Structure | R-work reported | R-free reported | R-work initial | R-free initial | R-work final | R-free final | Work CC initial | Free CC initial | Work CC final | Free CC final | Work CC Z-score | Free CC Z-score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2z81 | 0.213 | 0.232 | 0.2088 | 0.2224 | 0.1747 | 0.2135 | 0.9418 | 0.9381 | 0.9594 | 0.9435 | 31.30 | 1.84 |
| 2z81 rebuilt | N/A | N/A | 0.2056 | 0.2181 | 0.1733 | 0.2094 | 0.9437 | 0.9403 | 0.9602 | 0.9457 | 30.12 | 1.91 |
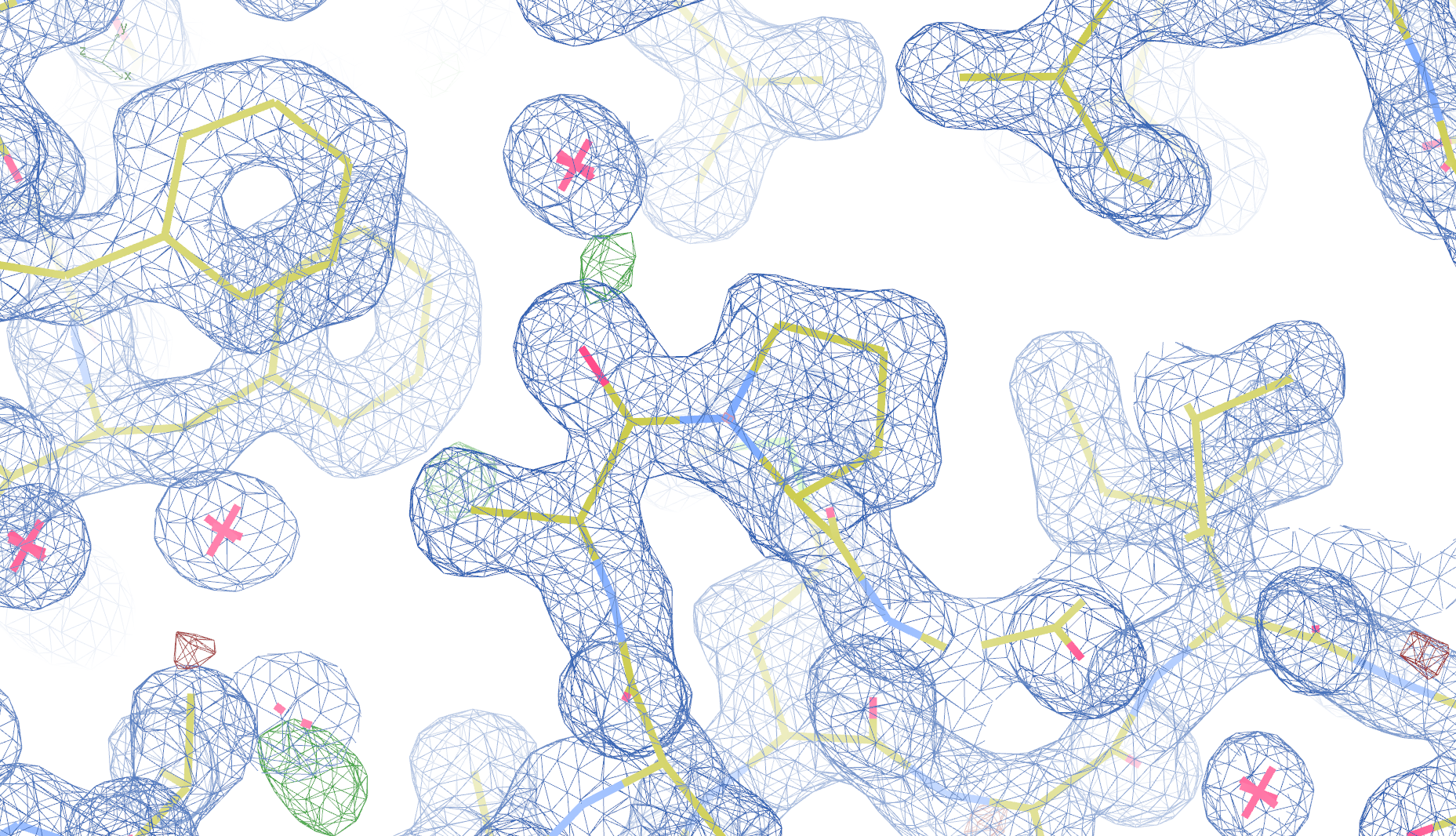
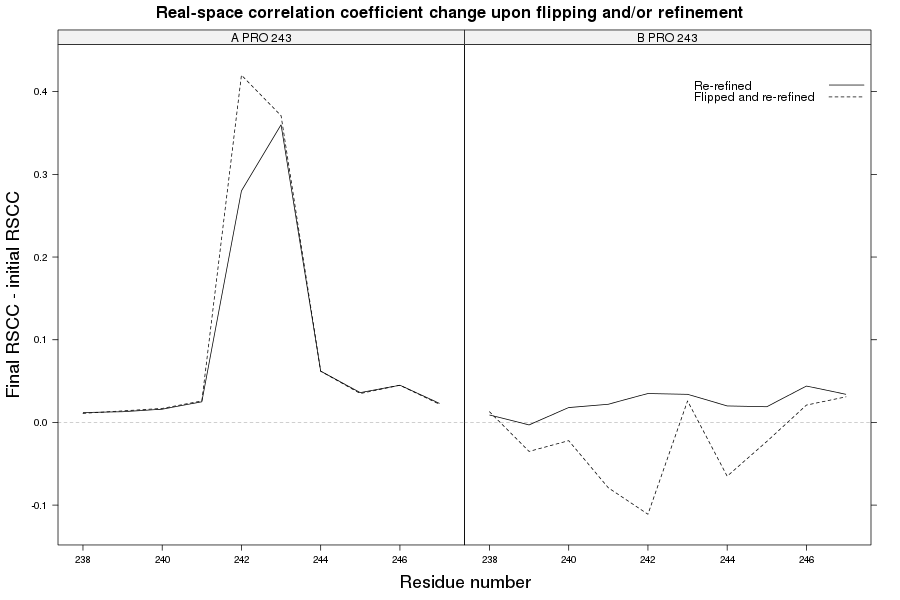
1pe9
The crystal structure of pectate lyase A 1pe9 has been solved at 1.60 Å and refined to reported R-/R-free factors of 0.198/0.213. 1 CISPEP record is present in the PDB file:
CISPEP 1 ALA B 242 PRO B 243 0 -0.11
However, there are two molecules of pectate lyase A in the asymmetric unit, and the same Ala-Pro dipeptide in needs to be flipped in chain A:
1pe9 PRO 243 - A tc+
These screenshots show both chains. There are still some difference
density peaks around B Pro 243, but they have vanished after
re-refinement.

The WHAT_CHECK validation report reports an unusually small Cδ-N-Cα angle for Pro 243 A and unusually short τ (N-Cα-C) angles for Ala 242 and Arg 244. Furthermore, the improper dihedral of the Pro 243 nitrogen deviates almost 10 σ from normal values, indicating distorted chirality. The puckering amplitude is also very high and the puckering phase is unusual. The torsion angles around Pro 243 are also unusual and the many bumps are present.
The following re-refinement statistics were obtained after 10 cycles of TLS refinement and 50 cycles restrained refinement in REFMAC with these parameters:
| Structure | R-work reported | R-free reported | R-work initial | R-free initial | R-work final | R-free final | Work CC initial | Free CC initial | Work CC final | Free CC final | Work CC Z-score | Free CC Z-score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1pe9 | 0.198 | 0.213 | 0.1906 | 0.2078 | 0.1619 | 0.1815 | 0.9482 | 0.9432 | 0.9598 | 0.9541 | 28.20 | 5.48 |
| 1pe9 rebuilt | N/A | N/A | 0.1894 | 0.2065 | 0.1612 | 0.1816 | 0.9489 | 0.9439 | 0.9601 | 0.9544 | 27.51 | 5.32 |