Introduction
Protein engineering, today, knows three major roads to success. So-called rational protein engineering approaches normally are based on a combination of common knowledge with protein structure analysis software, simulation techniques, and three dimensional visualisation. The second approach, the so-called evolutionary approaches are essentially a random mutagenesis followed by screening or selection ('directed evolution') of desired variants. The third approach relies on selection of proteins from the wide range of species for which the genome has been sequenced.
Rational engineering
So-called rational protein engineering approaches are based on a combination of common knowledge with information that either is extracted from the analysis of a multiple sequence alignment and, when available, the protein's structure or homology model. Energy based computational methods are often included and so is three dimensional visualisation and interaction. Rational methods have shown value for improving the stability of proteins, for redesigning parts of proteins, but their applicability in modifying the activity and specificity of enzymes has been limited.
Evolutionary methods
Numerous methods for library creation (i.e. error-prone PCR, mutator strains, DNA shuffling) and identification of the best hits (i.e. using phage technology, cell sorting by FACS, microtiterplate-based screening) have been developed in the past decade. One major limitation in directed evolution approaches is the huge library size due to the tremendous sequence space (for example, 20200 possible combinations for an enzyme composed of 200 amino acids). Researchers have tried to reduce the library size by applying focused directed evolution such as CASTing or ISM in which only a few positions are randomized simultaneously, but this can considerably affect the quality of the hits. The third approach relies on selection of proteins from the wide range of species for which the entire genome has been sequenced. This approach either involves direct selection of whole proteins, or the selected proteins can be used to extract ideas for mutations that can be applied to another protein. In addition, the recent 'omics' revolution has produced large numbers of miniaturisation and high throughput technologies that can be used well for the screening and selection that are integral aspects of evolutionary approaches.
Selection
Recently, the huge reservoir of (nucleotide or protein) sequences deposited in public databases derived from genome sequencing and metagenome sequencing opens the possibility for a novel approach in protein engineering: the use of bioinformatics tools to identify not only novel ˇV yet uncharacterized ˇV enzymes, but especially to identify differences or similarities in protein super-families. This information can be used to create highly specifically designed 'small-but-smart' libraries taking advantage of the effect that natural evolution over billions of years leads to the incorporation of beneficial mutations in the progeny of every organisms and hence in the proteins they produce. Thus, substantially smaller libraries have to be created and the success rate of finding hits ˇV improved biocatalysts, or more general proteins, with desired properties ˇV is substantially higher. The major key to make this approach working is the availability of easy to use bioinformatics tools to extract the required information from sequence databases.
State of the art, and beyond: software
In situations where the underlying theory is still immature researchers tend to opt for evolutionary or other random techniques. In case where the underlying concepts are well understood and the theory has been worked out with sufficient detail, rational approaches are often applied too. Active site engineering still is difficult from a theoretical point of view, so this class of protein engineering still relies largely on random approaches, and especially on random approaches that use small-but-smart libraries which design is guided by massive sequence analyses. Making a protein thermostable, on the other hand, is theoretically well-understood, and can often be approached through rational approaches.
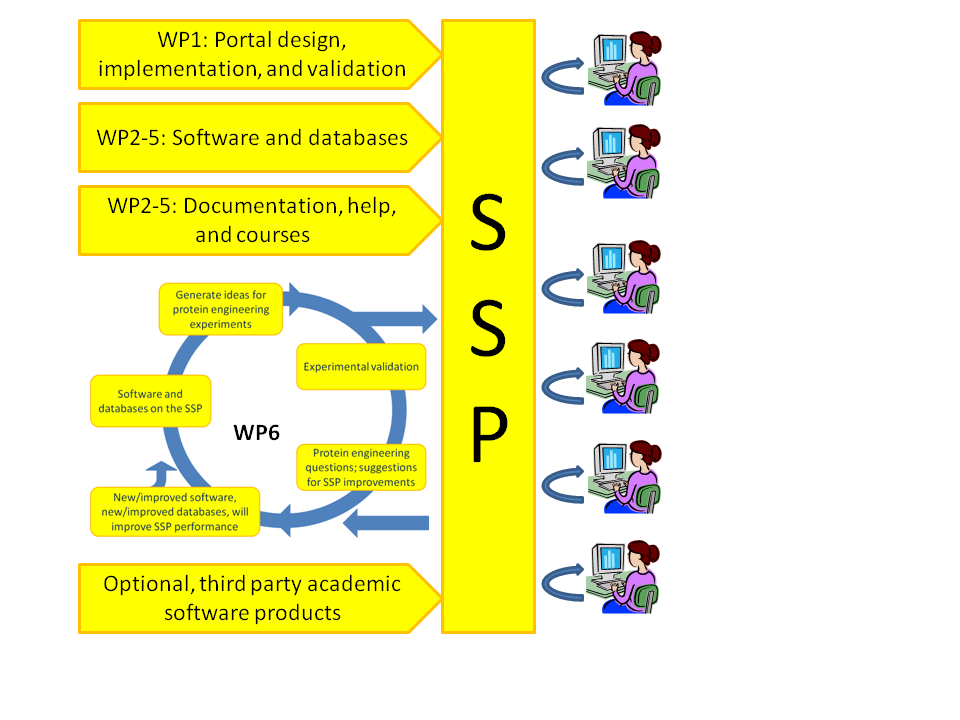 |
Proposal figure 1.1. Set-up of the NewProt project. The software partners
will design, implement, populate, and maintain the SSP self-service portal that
has an embedded interactive workbench. All predictive tools will be experimentally
validated. Users can interact with the portal in several interactive and user-friendly ways. |
Rational protein engineering approaches rely heavily on simulations and other computational techniques, and many computer programs have been described over the years that provide support in this field. Unfortunately, the aspiring protein engineer will need to struggle his way through a large series of software packages that either must be locally installed, or remotely executed on a large series of Web servers that all typically use different interfacing styles. To protein engineering SMEs this often provides a hurdle that is hard to take. The SSP will alleviate this problem by making available a large number of protein engineering facilities in a homogeneous, well-documented, and fully interoperable way. Additionally, all portal and workbench technology, software, and databases will be designed with server provisioning as a future possibility in mind. This will immediately make all facilities easily downloadable for researchers who prefer to do the work behind a firewall on their in-house computers, and will at a later stage
allow for much more powerful dissemination. Figure 1.1 indicates schematically how NewProtˇ¦s SSP is intended to carry biotechnology possibilities beyond the state of the art.
State of the art, and beyond: validation
In preparation for writing this proposal we analyzed all articles published in 2010 in the journal "Protein Engineering, Design, and Selection". We were struck by the fact that the materials and methods sections normally were rather extensive when dealing with the actual process of cloning, purification, and experimental testing, but that the paragraph on the bioinformatics aspects often consisted of only a few lines explaining which software was used to build the homology model or to select the locations where mutations could be made. Additionally, the results and conclusion section rarely reflected on the quality of the underlying software predictions. The NewProt partners, in contrast, will systematically experimentally validate the predictive power of the in silico protein engineering facilities through a series of activity, selectivity, and stability engineering experiments that will be performed in an iterative cycle with software optimisation. The software will suggest mutations and the validation will suggest software improvements.
In their selection of databases, software packages, tools, and information systems the NewProt partners will be guided by the WWW of protein engineering: Where, Which, and Why; Where should mutations be made; What mutations should be made; and Which mutations should be made at that position or those positions. The first two Ws, for Where and Which, will often come together, and are sufficient for most industrial applications. The third W, for Why, will be needed to efficiently go through the iterative steps of experimental validation and software and protocol improvement.
Innovation
The innovative aspects of the NewProt proposal include three main aspects:
- large series of existing, and newly designed in silico protein engineering facilities will be brought together in one, comprehensive, well documented, easy to use system that can be used freely and will be freely distributed;
- protein databases will be brought together, curated, and prepared for optimal use in the in silico protein engineering environment;
- the in silico protein engineering facilities will be tested experimentally, and the experimental results will both serve to improve the software methods and to illustrate the possibilities and limitations of these in silico protein engineering facilities.
The innovation potential of the NewProt proposal spreads far beyond the NewProt project and NewProt partners. A large series of FP7 calls will require access to good in silico protein engineering facilities to achieve the goals set by the Commission. These projects and their partners (academic and industrial alike) will be able to put the NewProt SSP to use to achieve their own goals in terms of innovation. The effects will be felt directly in the areas of protein engineering, biotechnology, and biocatalysis as new enzymes designed by this bioinformatics-driven approach will be created in the process of the experimental validation that will be marketed by the SMEs. Both biocatalysis companies hence have direct access to the most recent software tools required in modern protein engineering. They can apply these tools to their in-house developments and processes, and they can market the enzymes (variants) as well as produce chemicals by applying the biocatalysts available within the NewProt consortium. Neither of the experimental SMEs (Enzymicals, Ingenza) has the knowledge nor the capacity for such a bioinformatics approach. In turn, the bioinformatics SMEs learn from the academic and the experimental SMEs about the most important issues in the development of suitable biocatalysts and also profit from the partners knowledge within the consortium.