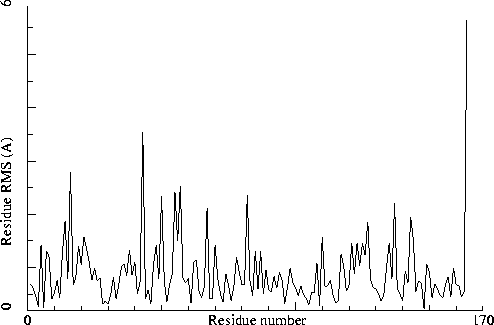
Chain identifiers of the two chains: A and B
All-atom RMS fit for the two chains : 1.105
CA-only RMS fit for the two chains : 0.508
Note: Non crystallographic symmetry backbone difference plot
The plot shows the differences in backbone torsion angles between
two similar chains on a residue-by-residue basis. Individual
"spikes" can be indicative of interesting or wrong residues. If all
residues show high differences, the structure could be incorrectly
refined.
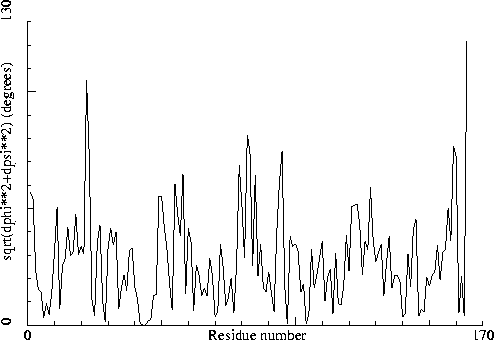
Chain identifiers of the two chains: A and B
Note: No crystallographic symmetry between molecules
No extra crystallographic symmetry was observed between the
independent molecules.
Note: Matthews coefficient OK
The Matthews coefficient [REF] is defined as the density of the protein structure in cubic Angstroms per Dalton. Normal values are between 1.5 (tightly packed, little room for solvent) and 4.0 (loosely packed, much space for solvent). Some very loosely packed structures can get values a bit higher than that.
Molecular weight of all polymer chains: 36996.816
Volume of the Unit Cell V= 2052018.9
Cell multiplicity: 18
Matthews coefficient for observed atoms Vm= 3.081
Note: All atoms are sufficiently far away from symmetry axes
None of the atoms in the structure is closer than 0.77 Angstrom to
a proper symmetry axis.
Sect 1) Administrative problems that can generate validation failures
Note: No strange inter-chain connections detected
No covalent bonds have been detected between molecules with
non-identical chain identifiers.
Note: No duplicate atom names in ligands
All atom names in ligands seem adequately unique.
Note: No mixed usage of alternate atom problems detected
Either this structure does not contain alternate atoms, or they have not
been mixed up, or the errors have remained unnoticed.
Note: In all cases the primary alternate atom was used
WHAT IF saw no need to make any alternate atom corrections (which means they
are all correct, or there arent any).
Note: No overlapping non-alternates detected
Either this structure does not contain overlapping non-alternate atoms,
or they are all correct, or the errors have remained unnoticed.
Note: No residues detected inside ligands
Either this structure does not contain ligands with amino acid groups
in it, or their naming is proper (enough).
Note: Ligand topologies OK
The topology could be determined for all ligands (or there are no ligands
for which a topology is needed, in which case there is absolutely no
problem, of course). That is good because it means that all ligands can
be included in the hydrogen bond optimization and related options.
Note: No attached groups interfere with hydrogen bond calculations
It seems there are no sugars, lipids, etc., bound (very close) to
atoms that otherwise could form hydrogen bonds.
Note: No atoms with high occupancy detected at special positions
Either there were no atoms at special positions, or all atoms at
special positions have adequately reduced occupancies.
Note: All residues have a complete backbone.
No residues have missing backbone atoms.
Note: No probable atoms with zero occupancy detected.
Either there are no atoms with zero occupancy, or they are not present in
the file, or their positions are sufficiently improbable to warrant a
zero occupancy.
Sect 2) Non-validating, descriptive output paragraph
Note: Content of the PDB file as interpreted by WHAT IF
Content of the PDB file as interpreted by WHAT IF
WHAT IF has read your PDB file, and stored it internally in
what is called 'the soup'. The content of this soup is listed here.
An extensive explanation of all frequently used WHAT IF output formats
can be found at http://swift.cmbi.umcn.nl/. Look under output formats.
1 1 ( 1) 164 ( 162) A Protein pdb104l.ent
2 165 ( 1) 328 ( 162) B Protein pdb104l.ent
3 329 ( HOH ) 329 ( HOH ) water ( 32) pdb104l.ent
In a colour picture, the residues that are part of a helix are shown in blue, strand residues in red. "Allowed" regions for helical residues are drawn in blue, for strand residues in red, and for all other residues in green.
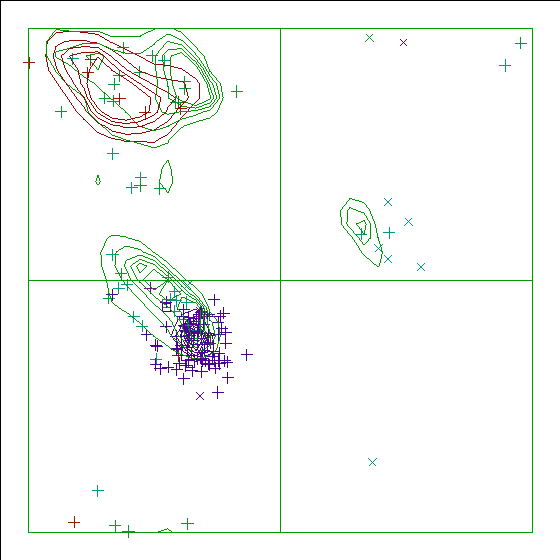
Chain identifier: A
Note: Ramachandran plot

Chain identifier: B
Note: Secondary structure
This is the secondary structure according to DSSP. Only helix (H),
overwound or 3/10-helix (3), strand (S), turn (T) and coil (blank)
are shown. [REF]. All DSSP related information can be found at
the DSSP page.
This is not really a structure validation option, but a very scattered
secondary structure (i.e. many strands of only a few residues length,
many Ts inside helices, etc) tends to indicate a poor structure.
10 20 30 40 50 60
| | | | | |
1 - 60 MNIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAKSAAELDKAIGRNTNGVI
( 1)-( 58) HHHHHHHHT T SS TTT SSSTTTSSSS TT HHHHHHHHHHHHTT TT
70 80 90 100 110 120
| | | | | |
61 - 120 TKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSL
( 59)-( 118) HHHHHHHHHHHHHHHHHHHTT TTTHHHHHHT HHHHHHHHHHHHHHHHHHHHT HHHH
130 140 150 160
| | | |
121 - 164 RMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYK
( 119)-( 162)HHTTTT HHHHHHHTTTTHHHHHTHHHHHHHHHHHHHTTT333
170 180 190 200 210 220
| | | | | |
165 - 224 MNIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSPSLNAAKSAAELDKAIGRNTNGVI
( 1)-( 58) HHHHHHHHH T SS TTT SS TTT TT HHHHHHHHHHHT T TT
230 240 250 260 270 280
| | | | | |
225 - 284 TKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSL
( 59)-( 118) HHHHHHHHHHHHHHHHHHHHT TTHHHHHHHT TTHHHHHHHHHHHH HHHHHH HHHH
290 300 310 320
| | | |
285 - 328 RMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYK
( 119)-( 162)HHHHTT TTHHHHHHTTTHHHHHT HHHHHHHHHHHT T3333
164 LYS ( 162 ) A 328 LYS ( 162 ) B
The distribution of the occupancy values in this file seems 'normal'.
Be aware that this evaluation is merely the result of comparing this
file with about 500 well-refined high-resolution files in the PDB. If
this file has much higher or much lower resolution than the PDB files
used in WHAT IF's trainig set, non-normal values might very well be
perfectly fine, or normal values might actually be not so normal...
Note: All occupancies seem to add up to 1.0.
In principle, the occupancy of all alternates of one atom should add up till
1.0. A valid exception is the missing atom (i.e. an atom not seen in the
electron density) that is allowed to have a 0.0 occupancy.
Warning: Average B-factor problem
The average B-factor for all buried protein atoms normally lies between
10--20. Values around 3--5 are expected for X-ray studies performed
at liquid nitrogen temperature.
Because of the extreme value for the average B-factor, no further analysis of the B-factors is performed.
Average B-factor for buried atoms : 43.737
Note: B-factor plot
The average atomic B-factor per residue is plotted as function of
the residue number.
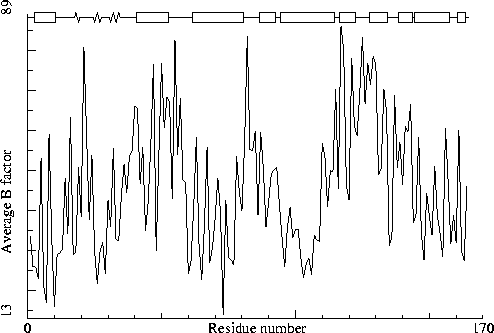
Chain identifier: A
Note: B-factor plot
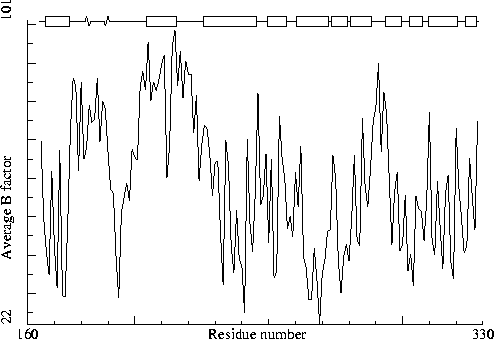
Chain identifier: B
Sect 4) Nomenclature related problems
Note: Valine nomenclature OK
No errors were detected in valine nomenclature.
Note: Threonine nomenclature OK
No errors were detected in threonine nomenclature.
Note: Isoleucine nomenclature OK
No errors were detected in isoleucine nomenclature.
Note: Leucine nomenclature OK
No errors were detected in leucine nomenclature.
Note: Arginine nomenclature OK
No errors were detected in arginine nomenclature.
Note: Tyrosine torsion conventions OK
No errors were detected in tyrosine torsion angle conventions.
Note: Phenylalanine torsion conventions OK
No errors were detected in phenylalanine torsion angle conventions.
Note: Aspartic acid torsion conventions OK
No errors were detected in aspartic acid torsion angle conventions.
Note: Glutamic acid torsion conventions OK
No errors were detected in glutamic acid torsion angle conventions.
Note: Heavy atom naming OK
No errors were detected in the atom names for non-hydrogen atoms.
Note: Chain names are OK
All chain names assigned to polymer molecules are unique, and all
residue numbers are strictly increasing within each chain.
Sect 5) Geometric checks
Warning: Unusual bond lengths
The bond lengths listed in the table below were found to deviate
more than 4 sigma from standard bond lengths (both standard values
and sigma for amino acid residues have been taken from Engh and
Huber [REF], for DNA they were taken from Parkinson et al [REF]). In
the table below for each unusual bond the bond length and the
number of standard deviations it differs from the normal value is
given.
Atom names starting with "-" belong to the previous residue in the chain. If the second atom name is "--SS", the disulphide bridge has a deviating length.
41 ALA ( 41 ) A N -C 1.230 -5.0 64 GLU ( 62 ) A CD OE2 1.325 4.0 66 GLU ( 64 ) A CD OE2 1.333 4.4 94 ASP ( 92 ) A CG OD2 1.334 4.5 110 GLU ( 108 ) A CA C 1.426 -4.7 130 GLU ( 128 ) A CD OE2 1.336 4.6 161 ASP ( 159 ) A CG OD1 1.332 4.4 227 ASP ( 61 ) B CG OD1 1.328 4.1 236 ASP ( 70 ) B CG OD2 1.334 4.4 274 GLU ( 108 ) B CD OE1 1.330 4.3 294 GLU ( 128 ) B CD OE2 1.330 4.2
RMS Z-score for bond lengths: 1.113
RMS-deviation in bond distances: 0.023
Warning: Possible cell scaling problem
Comparison of bond distances with Engh and Huber [REF] standard
values for protein residues and Parkinson et al [REF] values for
DNA/RNA shows a significant systematic deviation. It could be that
the unit cell used in refinement was not accurate enough. The
deformation matrix given below gives the deviations found: the
three numbers on the diagonal represent the relative corrections
needed along the A, B and C cell axis. These values are 1.000 in a
normal case, but have significant deviations here (significant at
the 99.99 percent confidence level)
There are a number of different possible causes for the discrepancy. First the cell used in refinement can be different from the best cell calculated. Second, the value of lambda used for a synchrotron data set can be miscalibrated. Finally, the discrepancy can be caused by a dataset that has not been corrected for significant anisotropic thermal motion.
Please note that the proposed scale matrix has NOT been constrained to obey the space group symmetry. This is done on purpose. The distortions can give you an indication of the accuracy of the determination.
Unit Cell deformation matrix
| 1.003013 0.000893 -0.001402| | 0.000893 1.002779 0.000555| | -0.001402 0.000555 1.001635|Proposed new scale matrix
| 0.005791 0.003341 0.000006| | -0.000006 0.006690 -0.000004| | 0.000017 -0.000007 0.012480|With corresponding cell
A = 172.606 B = 172.468 C = 80.131
Alpha= 89.865 Beta= 90.160 Gamma= 119.929
The CRYST1 cell dimensions
A = 172.100 B = 172.100 C = 80.000
Alpha= 90.000 Beta= 90.000 Gamma= 120.000
Variance: 72.753
(Under-)estimated Z-score: 6.286
Warning: Unusual bond angles
The bond angles listed in the table below were found to deviate
more than 4 sigma from standard bond angles (both standard values
and sigma for protein residues have been taken from Engh and Huber
[REF], for DNA/RNA from Parkinson et al [REF]). In the table below
for each strange angle the bond angle and the number of standard
deviations it differs from the standard values is given. Please
note that disulphide bridges are neglected. Atoms starting with "-"
belong to the previous residue in the sequence.
4 PHE ( 4 ) A CA CB CG 108.470 -5.3 6 MET ( 6 ) A N CA CB 102.973 -4.4 17 ILE ( 17 ) A CB CG1 CD1 101.793 -5.7 20 ASP ( 20 ) A CA CB CG 116.656 4.1 25 TYR ( 25 ) A CA CB CG 104.776 -4.6 31 HIS ( 31 ) A N CA CB 117.648 4.2 33 LEU ( 33 ) A -O -C N 130.061 4.4 33 LEU ( 33 ) A -CA -C N 107.355 -4.4 36 SER ( 36 ) A CA C O 113.665 -4.2 37 PRO ( 37 ) A -O -C N 128.068 4.3 37 PRO ( 37 ) A N CA CB 110.105 6.5 40 ASN ( 40 ) A CA CB CG 116.721 4.1 41 ALA ( 41 ) A -O -C N 133.563 6.6 41 ALA ( 41 ) A -CA -C N 105.509 -5.3 49 ASP ( 47 ) A CA CB CG 108.147 -4.5 56 THR ( 54 ) A CA CB OG1 102.421 -4.8 59 VAL ( 57 ) A CA CB CG1 102.581 -4.7 60 ILE ( 58 ) A -C N CA 113.663 -4.5 70 ASN ( 68 ) A CA CB CG 119.494 6.9 72 ASP ( 70 ) A N CA CB 121.198 6.3 72 ASP ( 70 ) A C CA CB 118.850 4.6 74 ASP ( 72 ) A CA CB CG 121.287 8.7 78 ARG ( 76 ) A CG CD NE 118.010 4.4 86 LEU ( 84 ) A N CA CB 117.433 4.1 91 ASP ( 89 ) A CA CB CG 107.361 -5.2And so on for a total of 69 lines.
RMS Z-score for bond angles: 1.565
RMS-deviation in bond angles: 2.973
Warning: Chirality deviations detected
The atoms listed in the table below have an improper dihedral value
that is deviating from expected values. As the improper dihedral values
are all getting very close to ideal values in recent X-ray structures,
and as we actually don't know how big the spread around these values
should be, this check only warns for 6 sigma deviations.
Improper dihedrals are a measure of the chirality/planarity of the structure at a specific atom. Values around -35 or +35 are expected for chiral atoms, and values around 0 for planar atoms. Planar side chains are left out of the calculations, these are better handled by the planarity checks.
Three numbers are given for each atom in the table. The first is the Z-score for the improper dihedral. The second number is the measured improper dihedral. The third number is the expected value for this atom type. A final column contains an extra warning if the chirality for an atom is opposite to the expected value.
3 ILE ( 3 ) A CA -6.1 22.7 34.1 72 ASP ( 70 ) A CA -10.5 13.4 34.0 118 ASN ( 116 ) A CA -10.8 13.3 34.2 122 MET ( 120 ) A CA 6.3 45.5 34.5 127 ARG ( 125 ) A CA -7.4 21.0 34.3 154 THR ( 152 ) A CA -6.6 22.1 34.3 172 ARG ( 8 ) B CA -8.3 19.5 34.3 180 LYS ( 16 ) B CA -6.6 23.1 34.3 195 HIS ( 31 ) B CA -7.5 21.8 34.7 230 GLU ( 64 ) B CA -7.1 23.2 34.4 290 LYS ( 124 ) B CA -11.5 14.9 34.3 292 TRP ( 126 ) B CA -6.1 24.6 34.7 309 PRO ( 143 ) B N -6.5 -40.7 -2.1 326 ALA ( 160 ) B CA -6.3 26.2 34.5
Improper dihedral RMS Z-score : 1.982
Error: Side chain planarity problems
The side chains of the residues listed in the table below contain a
planar group that was found to deviate from planarity by more than
4.0 times the expected value. For an amino acid residue that has a
side chain with a planar group, the RMS deviation of the atoms to a
least squares plane was determined. The number in the table is the
number of standard deviations this RMS value deviates from the
expected value (0.0).
227 ASP ( 61 ) B 4.534 267 ASN ( 101 ) B 4.490
37 PRO ( 37 ) A 0.19 LOW 88 PRO ( 86 ) A 0.13 LOW 252 PRO ( 86 ) B 0.17 LOW 309 PRO ( 143 ) B 0.18 LOW
145 PRO ( 143 ) A 4.5 envelop N (0 degrees)
These scores give an impression of how `normal' the torsion angles in protein residues are. All torsion angles except omega are used for calculating a `normality' score. Average values and standard deviations were obtained from the residues in the WHAT IF database. These are used to calculate Z-scores. A residue with a Z-score of below -2.0 is poor, and a score of less than -3.0 is worrying. For such residues more than one torsion angle is in a highly unlikely position.
117 THR ( 115 ) A -3.4044 182 TYR ( 18 ) B -2.8440 181 ILE ( 17 ) B -2.7865 218 ARG ( 52 ) B -2.7620 251 LYS ( 85 ) B -2.7304 3 ILE ( 3 ) A -2.7278 191 ILE ( 27 ) B -2.6888 185 THR ( 21 ) B -2.6664 9 ILE ( 9 ) A -2.6014 27 ILE ( 27 ) A -2.4852 216 ILE ( 50 ) B -2.4310 199 LYS ( 35 ) B -2.4022 198 THR ( 34 ) B -2.3370 34 THR ( 34 ) A -2.2955 128 TRP ( 126 ) A -2.2878 54 ARG ( 52 ) A -2.2532 224 ILE ( 58 ) B -2.2364 290 LYS ( 124 ) B -2.2050 266 ILE ( 100 ) B -2.2047 309 PRO ( 143 ) B -2.1999 175 GLU ( 11 ) B -2.1441 320 ARG ( 154 ) B -2.1258 316 ILE ( 150 ) B -2.1159 108 MET ( 106 ) A -2.1123 187 GLY ( 23 ) B -2.0862 50 LYS ( 48 ) A -2.0602 122 MET ( 120 ) A -2.0419 177 LEU ( 13 ) B -2.0414 321 THR ( 155 ) B -2.0251 257 LEU ( 91 ) B -2.0156 111 THR ( 109 ) A -2.0109
Residues with `forbidden' phi-psi combinations are listed, as well as residues with unusual omega angles (deviating by more than 3 sigma from the normal value). Please note that it is normal if about 5 percent of the residues is listed here as having unusual phi-psi combinations.
2 ASN ( 2 ) A Poor phi/psi 34 THR ( 34 ) A Poor phi/psi 37 PRO ( 37 ) A Poor PRO-phi 54 ARG ( 52 ) A Poor phi/psi 57 ASN ( 55 ) A Poor phi/psi 80 ILE ( 78 ) A Poor phi/psi 117 THR ( 115 ) A Poor phi/psi 126 LYS ( 124 ) A Poor phi/psi 145 PRO ( 143 ) A Poor PRO-phi 159 THR ( 157 ) A Poor phi/psi 181 ILE ( 17 ) B Poor phi/psi 182 TYR ( 18 ) B Poor phi/psi 187 GLY ( 23 ) B Poor phi/psi 191 ILE ( 27 ) B Poor phi/psi 198 THR ( 34 ) B Poor phi/psi 201 PRO ( 37 ) B Poor PRO-phi 202 SER ( 38 ) B Poor phi/psi 218 ARG ( 52 ) B Poor phi/psi 221 ASN ( 55 ) B Poor phi/psi 236 ASP ( 70 ) B Poor phi/psi 258 ASP ( 92 ) B Poor phi/psi 280 PHE ( 114 ) B Poor phi/psi 281 THR ( 115 ) B Poor phi/psi 290 LYS ( 124 ) B Poor phi/psi 309 PRO ( 143 ) B Poor phi/psi 321 THR ( 155 ) B Poor phi/psi 322 GLY ( 156 ) B Poor phi/psi 324 TRP ( 158 ) B Poor phi/psi
Ramachandran Z-score : -6.501
Warning: Omega angles too tightly restrained
The omega angles for trans-peptide bonds in a structure are
expected to give a gaussian distribution with the average around
+178 degrees and a standard deviation around 5.5 degrees. These
expected values were obtained from very accurately determined
structures. Many protein structures are too tightly constrained.
This seems to be the case with the current structure, as the
observed standard deviation is below 4.0 degrees.
Standard deviation of omega values : 0.511
Error: chi-1/chi-2 angle correlation Z-score very low
The score expressing how well the chi-1/chi-2 angles of all residues
are corresponding to the populated areas in the database is
very low.
chi-1/chi-2 correlation Z-score : -5.647
Warning: Backbone oxygen evaluation
The residues listed in the table below have an unusual backbone
oxygen position.
For each of the residues in the structure, a search was performed to find 5-residue stretches in the WHAT IF database with superposable C-alpha coordinates, and some constraints on the neighboring backbone oxygens.
In the following table the RMS distance between the backbone oxygen positions of these matching structures in the database and the position of the backbone oxygen atom in the current residue is given. If this number is larger than 1.5 a significant number of structures in the database show an alternative position for the backbone oxygen. If the number is larger than 2.0 most matching backbone fragments in the database have the peptide plane flipped. A manual check needs to be performed to assess whether the experimental data can support that alternative as well. The number in the last column is the number of database hits (maximum 80) used in the calculation. It is "normal" that some glycine residues show up in this list, but they are still worth checking!
321 THR ( 155 ) B 1.65 23 213 ASP ( 47 ) B 1.59 80 258 ASP ( 92 ) B 1.57 19
It is not necessarily an error if a few residues have rotamer values below 0.3, but careful inspection of all residues with these low values could be worth it.
167 ILE ( 3 ) B 0.37
For this check, backbone conformations are compared with database structures using C-alpha superpositions with some restraints on the backbone oxygen positions.
A residue mentioned in the table can be part of a strange loop, or there might be something wrong with it or its directly surrounding residues. There are a few of these in every protein, but in any case it is worth looking at!
29 ILE ( 29 ) A 0 160 TRP ( 158 ) A 0 163 TYR ( 161 ) A 0 164 LYS ( 162 ) A 0 165 MET ( 1 ) B 0 166 ASN ( 2 ) B 0 193 ILE ( 29 ) B 0 221 ASN ( 55 ) B 0 108 MET ( 106 ) A 1 118 ASN ( 116 ) A 1 137 LYS ( 135 ) A 1 191 ILE ( 27 ) B 2 216 ILE ( 50 ) B 2 220 THR ( 54 ) B 2 272 MET ( 106 ) B 2 301 LYS ( 135 ) B 2 324 TRP ( 158 ) B 2
Backbone conformation Z-score : -1.826
Sect 7) Bump checks
Error: Abnormally short interatomic distances
The pairs of atoms listed in the table below have an unusually
short distance.
The contact distances of all atom pairs have been checked. Two atoms are said to `bump' if they are closer than the sum of their Van der Waals radii minus 0.40 Angstrom. For hydrogen bonded pairs a tolerance of 0.55 Angstrom is used. The first number in the table tells you how much shorter that specific contact is than the acceptable limit. The second distance is the distance between the centers of the two atoms. Although we believe that two water atoms at 2.4 A distance are too close, we only report water pairs that are closer than this rather short distance.
The last text-item on each line represents the status of the atom pair. The text `INTRA' means that the bump is between atoms that are explicitly listed in the PDB file. `INTER' means it is an inter-symmetry bump. If the final column contains the text 'HB', the bump criterium was relaxed because there could be a hydrogen bond. Similarly relaxed criteria are used for 1--3 and 1--4 interactions (listed as 'B2' and 'B3', respectively). If the last column is 'BF', the sum of the B-factors of the atoms is higher than 80, which makes the appearance of the bump somewhat less severe because the atoms probably aren't there anyway.
Bumps between atoms for which the sum of their occupancies is lower than one are not reported. In any case, each bump is listed in only one direction.
87 LYS ( 85 ) A NZ -- 87 LYS ( 85 ) A NZ 0.863 2.137 INTER 40 ASN ( 40 ) A ND2 -- 43 LYS ( 43 ) A CE 0.736 2.364 INTRA BF 203 LEU ( 39 ) B CD2 -- 207 LYS ( 43 ) B CG 0.533 2.667 INTRA BF 78 ARG ( 76 ) A NE -- 98 ARG ( 96 ) A NH2 0.526 2.474 INTER BF 43 LYS ( 43 ) A NZ -- 301 LYS ( 135 ) B CB 0.487 2.613 INTRA BF 261 ARG ( 95 ) B NE -- 292 TRP ( 126 ) B CE3 0.372 2.728 INTRA BF 242 ARG ( 76 ) B NE -- 262 ARG ( 96 ) B NH1 0.356 2.644 INTER BF 201 PRO ( 37 ) B O -- 203 LEU ( 39 ) B N 0.346 2.354 INTRA BF 50 LYS ( 48 ) A CG -- 51 ALA ( 49 ) A N 0.334 2.766 INTRA BF 181 ILE ( 17 ) B N -- 191 ILE ( 27 ) B CD1 0.326 2.774 INTRA BF 316 ILE ( 150 ) B CG2 -- 317 THR ( 151 ) B N 0.324 2.776 INTRA BF 185 THR ( 21 ) B CG2 -- 186 GLU ( 22 ) B N 0.323 2.777 INTRA BF 180 LYS ( 16 ) B CG -- 181 ILE ( 17 ) B N 0.318 2.782 INTRA BF 173 ILE ( 9 ) B CG2 -- 174 ASP ( 10 ) B N 0.317 2.783 INTRA 59 VAL ( 57 ) A CG1 -- 60 ILE ( 58 ) A N 0.315 2.785 INTRA BF 232 LEU ( 66 ) B CA -- 235 GLN ( 69 ) B NE2 0.310 2.790 INTRA BF 111 THR ( 109 ) A CG2 -- 112 GLY ( 110 ) A N 0.309 2.791 INTRA BF 2 ASN ( 2 ) A CB -- 70 ASN ( 68 ) A ND2 0.307 2.793 INTER 190 THR ( 26 ) B CG2 -- 191 ILE ( 27 ) B N 0.305 2.795 INTRA BF 220 THR ( 54 ) B CG2 -- 221 ASN ( 55 ) B N 0.291 2.809 INTRA BF 323 THR ( 157 ) B O -- 325 ASP ( 159 ) B N 0.283 2.417 INTRA BF 317 THR ( 151 ) B O -- 320 ARG ( 154 ) B N 0.282 2.418 INTRA BF 308 THR ( 142 ) B CA -- 309 PRO ( 143 ) B CD 0.281 2.519 INTRA BF 113 VAL ( 111 ) A CG1 -- 114 ALA ( 112 ) A N 0.279 2.821 INTRA BF 10 ASP ( 10 ) A OD1 -- 150 ARG ( 148 ) A NH2 0.271 2.279 INTRA HBAnd so on for a total of 359 lines.
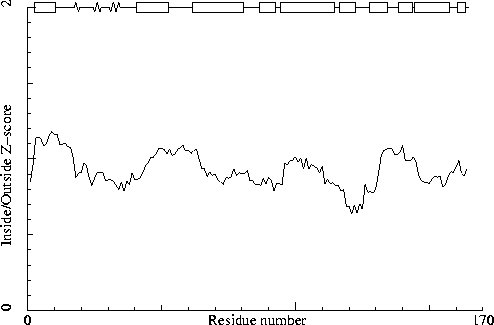
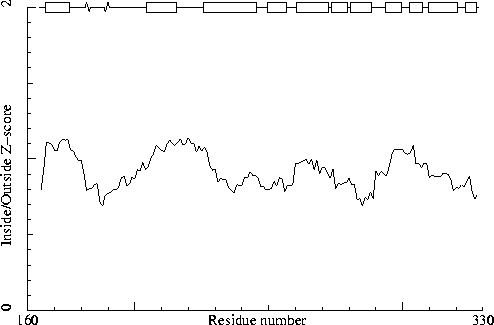
inside/outside RMS Z-score : 0.929
Note: Inside/Outside RMS Z-score plot
The Inside/Outside distribution normality RMS Z-score over a 15
residue window is plotted as function of the residue number. High
areas in the plot (above 1.5) indicate unusual inside/outside
patterns.
Chain identifier: A
Note: Inside/Outside RMS Z-score plot
Chain identifier: B
Warning: Abnormal packing environment for some residues
The residues listed in the table below have an unusual packing
environment.
The packing environment of the residues is compared with the average packing environment for all residues of the same type in good PDB files. A low packing score can indicate one of several things: Poor packing, misthreading of the sequence through the density, crystal contacts, contacts with a co-factor, or the residue is part of the active site. It is not uncommon to see a few of these, but in any case this requires further inspection of the residue.
246 ARG ( 80 ) B -6.48 199 LYS ( 35 ) B -5.67 143 GLN ( 141 ) A -5.61 307 GLN ( 141 ) B -5.55 35 LYS ( 35 ) A -5.52 271 GLN ( 105 ) B -5.37 107 GLN ( 105 ) A -5.29 156 ARG ( 154 ) A -5.26 320 ARG ( 154 ) B -5.26
Average for range 1 - 328 : -1.024
Note: Quality value plot
The quality value smoothed over a 10 residue window is plotted as
function of the residue number. Low areas in the plot (below
-2.0) indicate "unusual" packing.
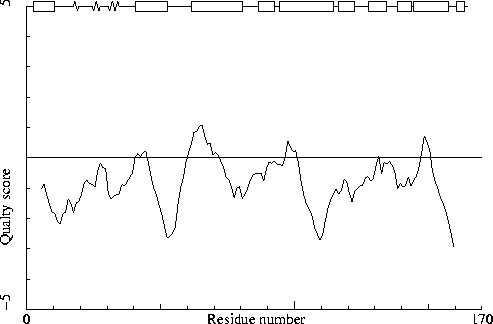
Chain identifier: A
Note: Quality value plot
The quality value smoothed over a 10 residue window is plotted as
function of the residue number. Low areas in the plot (below
-2.0) indicate "unusual" packing.
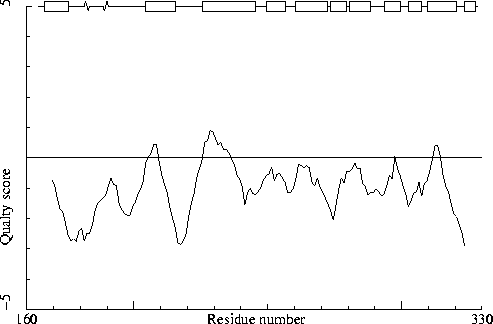
Chain identifier: B
Note: Second generation packing environment OK
None of the individual amino acid residues has a bad packing environment.
Note: No series of residues with abnormal new packing environment
There are no stretches of four or more residues each having a quality
control Z-score worse than -1.75.
Note: Structural average packing Z-score OK
The structural average for the second generation quality control
value is within normal ranges.
All contacts : Average = -0.177 Z-score = -0.80
BB-BB contacts : Average = -0.190 Z-score = -0.92
BB-SC contacts : Average = -0.018 Z-score = -0.16
SC-BB contacts : Average = -0.178 Z-score = -0.80
SC-SC contacts : Average = -0.111 Z-score = -0.35
Note: Second generation quality Z-score plot
The second generation quality Z-score smoothed over a 10 residue window
is plotted as function of the residue number. Low areas in the plot (below
-1.3) indicate "unusual" packing.
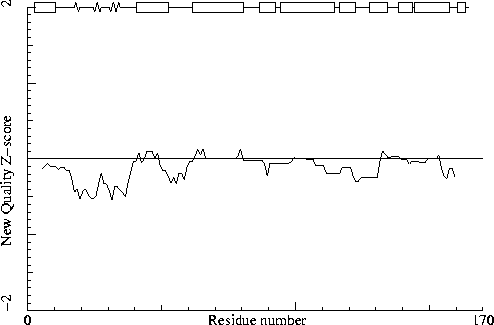
Chain identifier: A
Note: Second generation quality Z-score plot
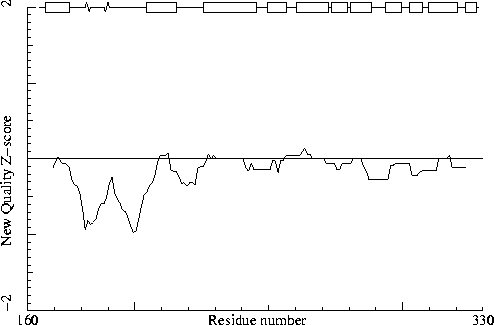
Chain identifier: B
Sect 9) Water and hydrogenbond related checks
Error: Water clusters without contacts with non-water atoms
The water molecules listed in the table below are part of water
molecule clusters that do not make contacts with non-waters. These
water molecules are part of clusters that have a distance at least
1 Angstrom larger than the sum of the Van der Waals radii to the
nearest non-solvent atom. Because these kinds of water clusters
usually are not observed with X-ray diffraction their presence
could indicate a refinement artifact. The number in brackets is the
identifier of the water molecule in the input file.
329 HOH ( 11 ) 329 HOH ( 12 )
The number in brackets is the identifier of the water molecule in the input file. Suggested coordinates are also given in the table. Please note that alternative conformations for protein residues are not taken into account for this calculation. If you are using WHAT IF / WHAT-CHECK interactively, then the moved waters can be found in PDB format in the file: MOVED-H2O.pdb.
329 HOH ( 11 ) -27.543 43.420 17.460 329 HOH ( 12 ) -27.940 36.120 31.439 329 HOH ( 16 ) -7.833 15.429 41.757 329 HOH ( 27 ) -21.772 39.782 38.071
329 HOH ( 5 ) 329 HOH ( 11 ) 329 HOH ( 12 )
55 ASN ( 53 ) A 221 ASN ( 55 ) B
In the table below all normal histidine residues are listed. The assignment based on the geometry of the residue is listed first, together with the RMS Z-score for the fit to the Engh and Huber parameters. For all residues where the H-bond assignment is different, the assignment is listed in the last columns, together with its RMS Z-score to the Engh and Huber parameters.
As always, the RMS Z-scores should be close to 1.0 if the residues were restrained to the Engh and Huber parameters during refinement.
Please note that because the differences between the geometries of the different types are small it is possible that the geometric assignment given here does not correspond to the type used in refinement. This is especially true if the RMS Z-scores are much higher than 1.0.
If the two assignments differ, or the `geometry' RMS Z-score is high, it is advisable to verify the hydrogen bond assignment, check the HIS type used during the refinement and possibly adjust it.
31 HIS ( 31 ) A HIS-D 1.04 195 HIS ( 31 ) B HIS-D 0.31
Hydrogen bond donors that are buried inside the protein normally use all of their hydrogens to form hydrogen bonds within the protein. If there are any non hydrogen bonded buried hydrogen bond donors in the structure they will be listed here. In very good structures the number of listed atoms will tend to zero.
Waters are not listed by this option.
2 ASN ( 2 ) A N 12 GLY ( 12 ) A N 22 GLU ( 22 ) A N 25 TYR ( 25 ) A N 25 TYR ( 25 ) A OH 34 THR ( 34 ) A N 38 SER ( 38 ) A N 40 ASN ( 40 ) A N 40 ASN ( 40 ) A ND2 41 ALA ( 41 ) A N 94 ASP ( 92 ) A N 103 ASN ( 101 ) A ND2 126 LYS ( 124 ) A N 140 TRP ( 138 ) A N 160 TRP ( 158 ) A N 160 TRP ( 158 ) A NE1 166 ASN ( 2 ) B N 178 ARG ( 14 ) B N 186 GLU ( 22 ) B N 189 TYR ( 25 ) B N 202 SER ( 38 ) B OG 203 LEU ( 39 ) B N 204 ASN ( 40 ) B N 222 GLY ( 56 ) B N 262 ARG ( 96 ) B N 262 ARG ( 96 ) B NH1 265 LEU ( 99 ) B N 312 ALA ( 146 ) B N 318 THR ( 152 ) B OG1
Side-chain hydrogen bond acceptors that are buried inside the protein normally form hydrogen bonds within the protein. If there are any not hydrogen bonded in the optimized hydrogen bond network they will be listed here.
Waters are not listed by this option.
306 ASN ( 140 ) B OD1
The second part of the table mostly gives an impression of how well the model conforms to common refinement constraint values. The first part of the table shows a number of constraint-independent quality indicators.
Structure Z-scores, positive is better than average:
1st generation packing quality : -1.310 2nd generation packing quality : -0.796 Ramachandran plot appearance : -6.501 (bad) chi-1/chi-2 rotamer normality : -5.647 (bad) Backbone conformation : -1.826
Bond lengths : 1.113 Bond angles : 1.565 Omega angle restraints : 0.093 (tight) Side chain planarity : 1.464 Improper dihedral distribution : 1.982 (loose) Inside/Outside distribution : 0.929
The second part of the table mostly gives an impression of how well the model conforms to common refinement constraint values. The first part of the table shows a number of constraint-independent quality indicators, which have been calibrated against structures of similar resolution.
Resolution found in PDB file : 2.80
Structure Z-scores, positive is better than average:
1st generation packing quality : -0.3 2nd generation packing quality : 0.8 Ramachandran plot appearance : -3.9 (poor) chi-1/chi-2 rotamer normality : -3.2 (poor) Backbone conformation : -1.2
Bond lengths : 1.113 Bond angles : 1.565 Omega angle restraints : 0.093 (tight) Side chain planarity : 1.464 Improper dihedral distribution : 1.982 (loose) Inside/Outside distribution : 0.929 ==============
WHAT IF
G.Vriend,
WHAT IF: a molecular modelling and drug design program,
J. Mol. Graph. 8, 52--56 (1990).
WHAT_CHECK (verification routines from WHAT IF)
R.W.W.Hooft, G.Vriend, C.Sander and E.E.Abola,
Errors in protein structures
Nature 381, 272 (1996).
Bond lengths and angles, protein residues
R.Engh and R.Huber,
Accurate bond and angle parameters for X-ray protein structure
refinement,
Acta Crystallogr. A47, 392--400 (1991).
Bond lengths and angles, DNA/RNA
G.Parkinson, J.Voitechovsky, L.Clowney, A.T.Bruenger and H.Berman,
New parameters for the refinement of nucleic acid-containing structures
Acta Crystallogr. D52, 57--64 (1996).
DSSP
W.Kabsch and C.Sander,
Dictionary of protein secondary structure: pattern
recognition of hydrogen bond and geometrical features
Biopolymers 22, 2577--2637 (1983).
Hydrogen bond networks
R.W.W.Hooft, C.Sander and G.Vriend,
Positioning hydrogen atoms by optimizing hydrogen bond networks in
protein structures
PROTEINS, 26, 363--376 (1996).
Matthews' Coefficient
B.W.Matthews
Solvent content of Protein Crystals
J. Mol. Biol. 33, 491--497 (1968).
Protein side chain planarity
R.W.W. Hooft, C. Sander and G. Vriend,
Verification of protein structures: side-chain planarity
J. Appl. Cryst. 29, 714--716 (1996).
Puckering parameters
D.Cremer and J.A.Pople,
A general definition of ring puckering coordinates
J. Am. Chem. Soc. 97, 1354--1358 (1975).
Quality Control
G.Vriend and C.Sander,
Quality control of protein models: directional atomic
contact analysis,
J. Appl. Cryst. 26, 47--60 (1993).
Ramachandran plot
G.N.Ramachandran, C.Ramakrishnan and V.Sasisekharan,
Stereochemistry of Polypeptide Chain Conformations
J. Mol. Biol. 7, 95--99 (1963).
Symmetry Checks
R.W.W.Hooft, C.Sander and G.Vriend,
Reconstruction of symmetry related molecules from protein
data bank (PDB) files