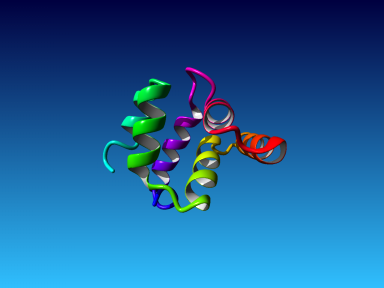
The three-dimensional structure of the following target sequence has been predicted by YASARA's homology modeling experiment, Version 20.7.4.L.64:
>IRAKM LLFDLPPALLGELCAVLDSCDGALGWRGLAERLSSSWLDVRHIEKYVDQGKSGTRELLWS WAQKNKTIGDLLQVLQEMGHRRAIHLITNY
The target sequence contains 90 residues in 1 molecule.
The following parameters have been chosen for this target:
The following 2 template structures were provided directly:
| Template | Object | Name | Resolution | Amino acids |
| 1 | 1 | T001 | Unknown or NMR | 88 |
| 2 | 2 | T002 | Unknown or NMR | 89 |
Since an alignment has been provided, there is no need for a secondary structure prediction, which was therefore skipped to save time. Instead, equal probabilities have been assigned to all secondary structure types. The resulting prediction is listed below, the lines 'PreHel', 'PreStr' and 'PreCoi' indicate the estimated probability for the three secondary structure classes helix, strand and coil.
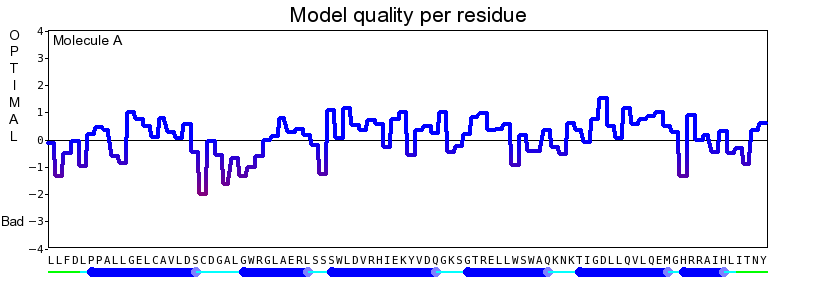
Sequence: LLFDLPPALLGELCAVLDSCDGALGWRGLAERLSSSWLDVRHIEKYVDQGKSGTRELLWSWAQKNKTIGDLLQVLQEMGHRRAIHLITNY SecStr : CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC PreHel : 333333333333333333333333333333333333333333333333333333333333333333333333333333333333333333 PreStr : 333333333333333333333333333333333333333333333333333333333333333333333333333333333333333333 PreCoi : 333333333333333333333333333333333333333333333333333333333333333333333333333333333333333333
For each of the templates listed above, one model was built based on the alignment specified in /home/user/projects/IRAK/modeling/model18/irakm_align.fasta.
This model is a monomer, and based on the following user-provided sequence alignment:
Target: LLFDLPPALLGELCAVLDSCDGALGWRGLAERLSSSWLDVRHIEKYVDQGKSGTRELLWSWAQKNKTIGDLLQVLQEMGHRRAIHLITNY Match: | LP : :|LCA LD: W: LA :: :V::I:: ::G|S:::E:L W Q N T| L: ::::| ::A::LI :Y Template:AIRLLPLPVRAQLCAHLDA.....VWQQLATAVKLYPDQVEQISSQKQRGRSASNEFLNIW.QYNHTVQTLFALFKKLKLHNAMRLIKDY SecStr: CCCCCHHHHHHHHHHHHHH.....HHHHHHHHHCCHHHHHHHHHHHHHCCCHHHHHHHHHH.HCCCHHHHHHHHHHHHCHHHHHHHCCCC
In the alignment above, 84 of 90 target residues (93.3%) are aligned to template residues. Among these aligned residues, the sequence identity is 29.8% and the sequence similarity is 48.8% ('similar' means that the BLOSUM62 score is > 0). The following 2 loops had to be modeled:
| Loop | N-terminal anchor | Loop sequence | C-terminal anchor |
| 1 | LLWSW | AQ | KNKTI |
| 2 | AVLDS | CDGAL | GWRGL |
After the side-chains had been built, optimized and fine-tuned, all newly modeled parts were subjected to a combined steepest descent and simulated annealing minimization (i.e. the backbone atoms of aligned residues were kept fixed to avoid potential damage).
The resulting half-refined model has been saved as irakm_t001-a_refined050.yob and obtained the following quality Z-scores:
| Check type | Quality Z-score | Comment |
| Dihedrals | 0.549 | Optimal |
| Packing 1D | -0.928 | Good |
| Packing 3D | 0.025 | Optimal |
| Overall | -0.271 | Good |
Then a full unrestrained simulated annealing minimization was run for the entire model. The result has been saved as irakm_t001-a_refined100.yob, the corresponding Z-scores are listed below:
| Check type | Quality Z-score | Comment |
| Dihedrals | 0.804 | Optimal |
| Packing 1D | -0.475 | Good |
| Packing 3D | 0.141 | Optimal |
| Overall | -0.003 | Good |
Since the overall quality Z-score improved to -0.003 during the minimization, this fully refined model has been accepted as the final one for this template and alignment.
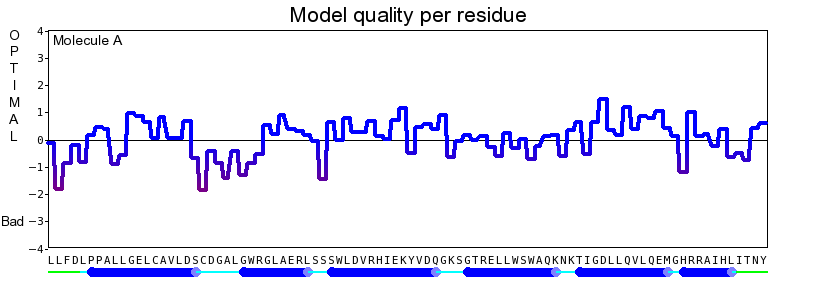
The final model for this template and alignment has been saved as irakm_t001-a.yob, and is shown below, together with a plot of its overall quality Z-score, shown per residue:
| |
This model is a monomer, and based on the following user-provided sequence alignment:
Target: LLFDLPPALLGELCAVLDSCDGALGWRGLAERLSSSWLDVRHIEKYVDQGKSGTRELLWSWAQKNKTIGDLLQVLQEMGHRRAIHLITNY Match: | :L :|L :L |D | LA | : : |R::E G S T ELL::W: N T|GDL:::L |: L| Template:YIRNLNVGILRKLSDFID........KKLAVAI..NQFHIRRFEAL..TGLSPTCELLFDWGTTNCTVGDLVDLLVQIELFAPATLLL.. SecStr: CCCCCHHHHHHHHHHHHC........HHHHHHE..HHHHHHHHHHC..CCCHHHHHHHHHHHCCCCHHHHHHHHHHHHCCHHHHHHHH..
In the alignment above, 76 of 90 target residues (84.4%) are aligned to template residues. Among these aligned residues, the sequence identity is 28.9% and the sequence similarity is 50.0% ('similar' means that the BLOSUM62 score is > 0). The following 4 loops had to be modeled:
| Loop | N-terminal anchor | Loop sequence | C-terminal anchor |
| 1 | LAERL | SSSW | LDVRH |
| 2 | HIEKY | VD | QGKSG |
| 3 | CAVLD | SCDGALGW | RGLAE |
| 4 | IHLIT | NY | None |
After the side-chains had been built, optimized and fine-tuned, all newly modeled parts were subjected to a combined steepest descent and simulated annealing minimization (i.e. the backbone atoms of aligned residues were kept fixed to avoid potential damage).
The resulting half-refined model has been saved as irakm_t002-a_refined050.yob and obtained the following quality Z-scores:
| Check type | Quality Z-score | Comment |
| Dihedrals | -0.042 | Good |
| Packing 1D | -1.344 | Satisfactory |
| Packing 3D | -0.493 | Good |
| Overall | -0.760 | Good |
Then a full unrestrained simulated annealing minimization was run for the entire model. The result has been saved as irakm_t002-a_refined100.yob, the corresponding Z-scores are listed below:
| Check type | Quality Z-score | Comment |
| Dihedrals | 0.173 | Optimal |
| Packing 1D | -0.736 | Good |
| Packing 3D | -0.455 | Good |
| Overall | -0.473 | Good |
Since the overall quality Z-score improved to -0.473 during the minimization, this fully refined model has been accepted as the final one for this template and alignment.
NOTE: 2 residues contained in N- and C-terminal unaligned tails of the monomeric model have been excluded from the Z-scores in both tables above, since they are not reliable (YASARA does not yet perform ab initio structure prediction) and would have mainly added noise to the result.
The final model for this template and alignment has been saved as irakm_t002-a.yob, and is shown below, together with a plot of its overall quality Z-score, shown per residue:
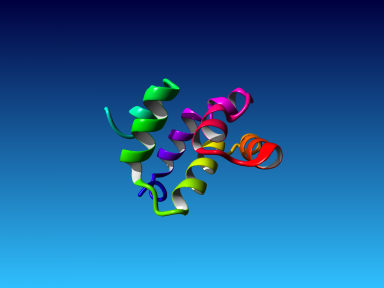 | 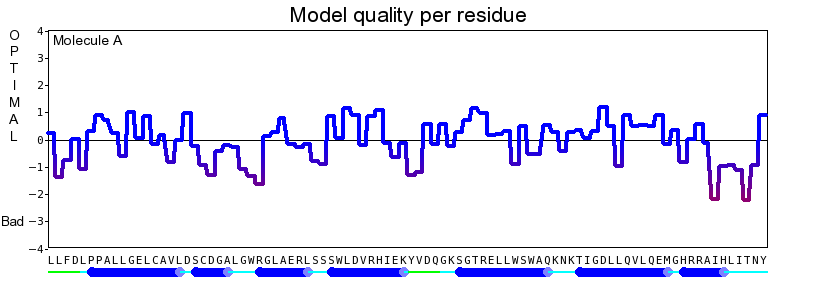 |
The following table lists the 2 models sorted by their overall quality Z-scores. The models have been superposed and saved together as irakm.sce.
| Rank | Z-score | Structure | State | Model ID | Filename | Original number | Residues | Comment |
| 1 | -0.003 | monomer | T001-A | irakm_t001-a.yob | 1 | 1-90 | Good | |
| 2 | -0.473 | monomer | T002-A | irakm_t002-a.yob | 2 | 1-88 | Good |
Finally, YASARA tried to combine the best parts of the 2 models to obtain a hybrid model, hoping to increase the accuracy beyond each of the contributors. The following fragments were copied from other models (note that the first transfer is simply the initial model considered most suitable for hybridization, and that the scores in the right column are not comparable to the Z-scores listed further above, since they now penalize missing and very exposed residues):
| Transfer | First residue | Last residue | Length | From model | Score |
| 1 | 1 | 90 | 90 | T001-A | -1.466 |
| 2 | 52 | 63 | 12 | T002-A | -1.385 accepted |
The resulting hybrid model obtained the following quality Z-scores (this time the score includes floppy terminal tails):
| Check type | Quality Z-score | Comment |
| Dihedrals | 0.733 | Optimal |
| Packing 1D | 0.070 | Optimal |
| Packing 3D | 0.165 | Optimal |
| Overall | 0.210 | Optimal |
The following figure shows the initial model in blue, and all hybridized parts in a different color.
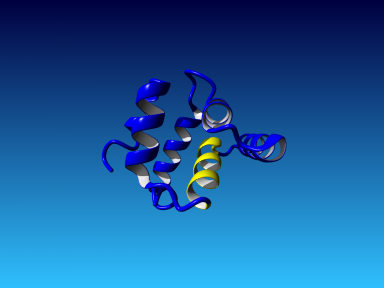 |  |
This hybrid model with Z-score 0.210 was saved as the final one, irakm.yob
NOTE: If the hybrid model scores worse than the model from which it was initially derived, this is not necessarily a bad thing, since the hybrid model often covers more residues.
NOTE: A Z-score describes how many standard deviations the model quality is away from the average high-resolution X-ray structure. Higher values are better, negative values indicate that the homology model looks worse than a high-resolution X-ray structure. The overall Z-scores for all models have been calculated as the weighted averages of the individual Z-scores using the formula Overall = 0.145*Dihedrals + 0.390*Packing1D + 0.465*Packing3D. The overall score thus captures the correctness of backbone- (Ramachandran plot) and side-chain dihedrals, as well as packing interactions. It applies to globular proteins only, and can be mislead by artificial structures like long single alpha helices (which have perfect dihedrals and are free of packing errors, since there is no packing).
Created Mon Aug 31 12:40:54 2020 by YASARA Structure.