Administrative
Some of the 20 amino acid types in proteins have atoms that "look" the
same. So, for instance, the Cδ1 and Cδ2 atoms in phenylalanine
look the same, and thus all phenylalanine residues would have two ways of naming the
atoms:
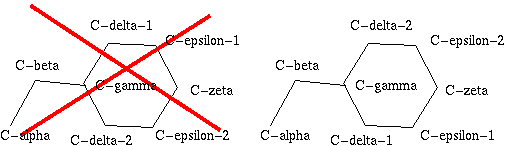
|
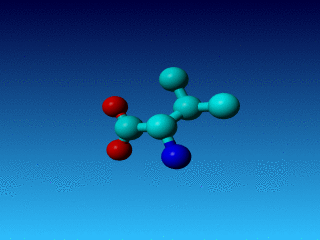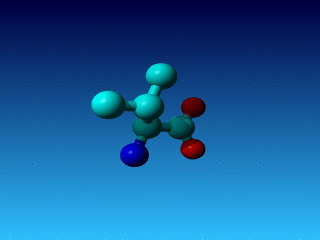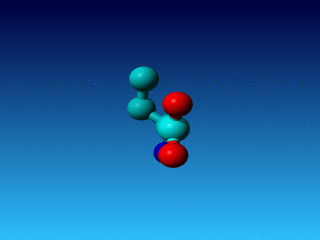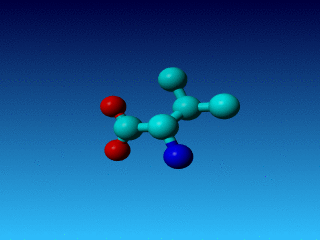
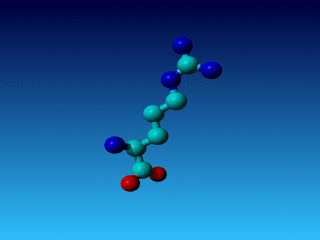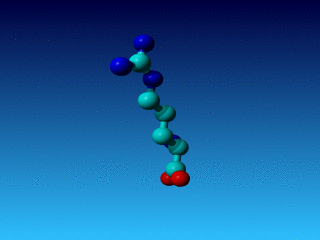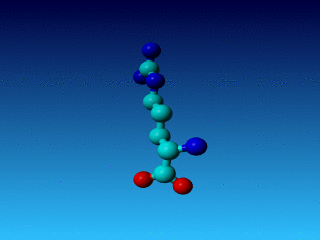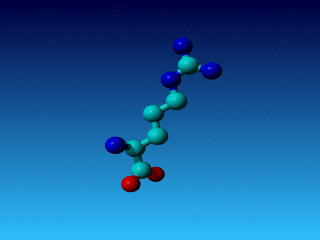
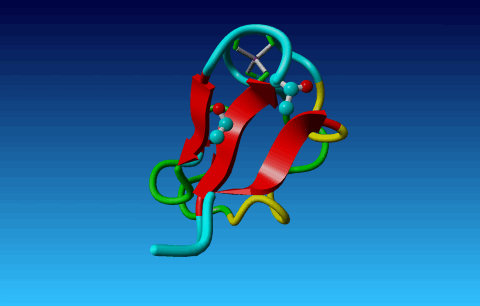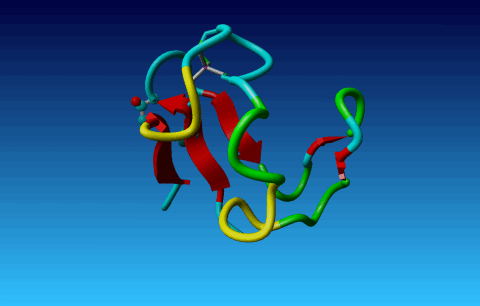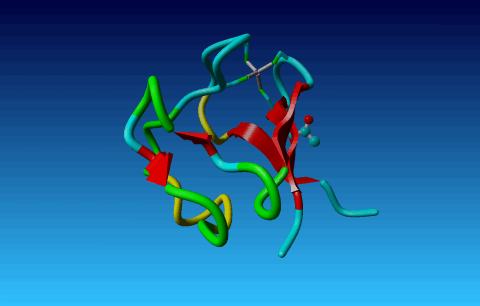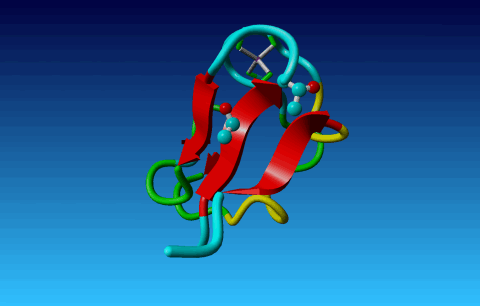
Figure 24. Phe has two seemingly equivalent rotamers. Biophysically, these
two rotamers are the same. But for a bioinformatician they can be different.
Just think of superposing two structures with software that isn't smart
enough to see that the two rotamers are equivalent...
|
Similar problems can occur in a series of residues, and WHAT_CHECK warns for
all of them:
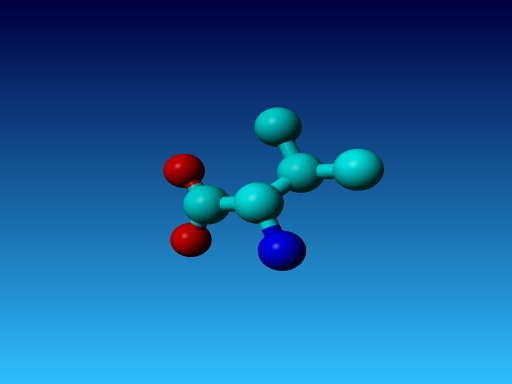
|
Figure 25. In Val the Cγ1 and Cγ2 cannot be swapped.
|
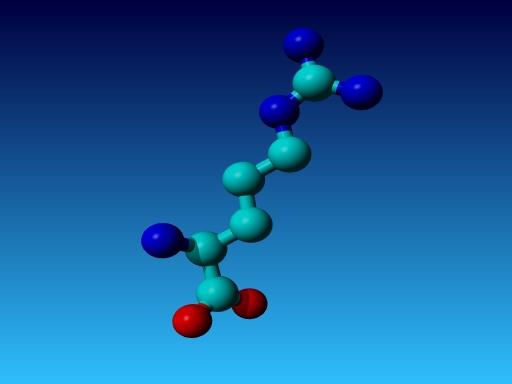
|
Figure 26. In Arg the Cγ1 and Cγ2 cannot be swapped.
|
Swappable atoms
Similar problems can occur in a series of residues in which atoms (or atom names)
can be found swapped without there being any actual biophysical error (but it
seems wise to correct these swapped situations anyway):
| Residue |
Swappable atoms |
| Asp |
Oδ1 and Oδ2 |
| Glu |
Oε1 and Oε2 |
| Phe |
Cδ1 and Cδ2; Cε1 and Cε2 |
| Ile |
Cγ1 and Cγ2 |
| Leu |
Cδ1 and Cδ2; |
| Val |
Cγ1 and Cγ2 |
| Tyr |
Cδ1 and Cδ2; Cε1 and Cε2 |
In all these cases there is nothing wrong biophysically speaking. But as such
swaps make it difficult to optimally use the coordinates in (in silico) follow-up
studies a correction would be nice.
Swaps that are worth an error message instead of a warning
Obviously, if for example, the Cδ1 and Cδ2 have been swapped
in phenylalanine but the Cε1 and Cε2 have not been swapped then
we still did not disobey any law of physics, but it gets even harder to use
the coordinates sensibly in drug design, in protein engineering, etc.
In this section we show some swaps that actually constitute errors, or get very close
to being a real error.
Isoleucine
|
Figure 27. If the Cγ1 and Cγ2 are swapped in isoleucine, then
many softwares will draw strange bonds. Like here residue 318 in PDB file 1ARX.
In this case there still nothing wrong, biophysically speaking.
|
|
Figure 28. But in these two isoleucines, in PDB file 1AZE, the error is more
than just administrative. these two isoleucines are also biophysically wrong.
|
Threonine

|
Figure 29. In Thr the Oγ is called Oγ1 and the Cγ
is called Cγ2. So it is possible to make two kinds of errors here:
Numbering the C and O the wrong way around, and actually swapping them. The latter
is discussed below.
|
|
Figure 30. However, with threonine things can also go very, very wrong.
In this 1.2 Ångström resolution structure two threonines are observed very
close to each other, with opposite hands on their Cβ.
|
We contacted the depositors of this PDB file (5RXN) about the funny Thr. To show
how difficult things can be, these authors wrote us back that "both threonines
were nicely located in good density, so they don't see anything wrong and don't
know what we were talking about".
Histidine
In histidine we see a similar problem as in threonine. The δ-atoms should
be called Nδ1 and Cδ2. Consequently, following the rules in organic
chemistry for atom naming further, the ε-atoms should be called
Cε1 and Nε2. Now two kinds of errors can be made. The first
is swapping the names. This is inconvenient for the users of the coordinates.
However, swapping the atoms for either only the δ-atoms, or only
the ε-atoms makes it a serious error as is illustrated with a picture
from one example found in the PDB file 1HCE.
|
Figure 31. In Histidine with the Nδ1 and the Cδ2 swapped (or
the ε-atoms swapped, of course; that difference cannot be seen).
|
WHAT_CHECK reacts 'shocked' on this latter case. It reports weird bond
lengths (details of the numbers in these tables are discussed later on in
this WHAT_CHECK user course):
27 HIS ( 27-) A - CB CG 1.68 13.3
27 HIS ( 27-) A - CG ND1 1.48 6.4
27 HIS ( 27-) A - CG CD2 1.52 15.2
27 HIS ( 27-) A - ND1 CE1 1.41 7.0
|
It reports weird bond angles:
27 HIS ( 27-) A - CG ND1 CE1 32.66 -72.9
27 HIS ( 27-) A - ND1 CE1 NE2 26.96 -65.2
27 HIS ( 27-) A - CE1 NE2 CD2 41.22 -50.5
27 HIS ( 27-) A - NE2 CD2 CG 43.89 -62.6
27 HIS ( 27-) A - CD2 CG ND1 35.53 -70.6
27 HIS ( 27-) A - CB CG CD2 154.10 19.2
|
It reports bumps:
27 HIS ( 27-) A - CG <--> 28 HIS ( 28-) A - N 0.32 2.68 INTRA
27 HIS ( 27-) A - CE1 <--> 28 HIS ( 28-) A - CD2 0.28 2.92 INTRA
|
And it reports a weird packing environment:
27 HIS ( 27-) A - --- 31 HIS ( 31-) A - -5.37
|
Atom names
The names of the atoms in most residues have been fixed by the PDB. The concept
originally was that the atom in the main chain is called the α-atom; and
for most residues -certainly the 20 canonical ones- that is then the Cα.
The first atom into the side chain is called the β atom, etcetera. As it was
not possible in the very early days of the PDB to use Greek characters in
computers, it was decided to use the European equivalents of the Greek
characters.
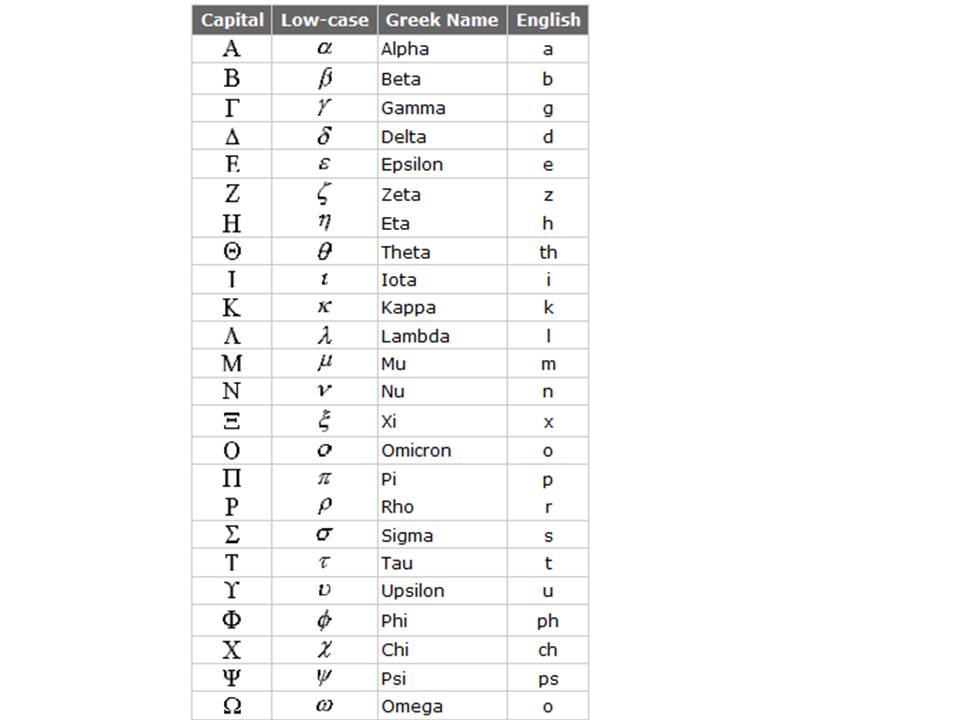
|
Figure 32. The Greek alphabet with the European equivalent characters indicated.
(Figure obtained from http://www.physlink.com/reference/greekalphabet.cfm).
|
Unfortunately, the PDB doesn't always stick to this rule...
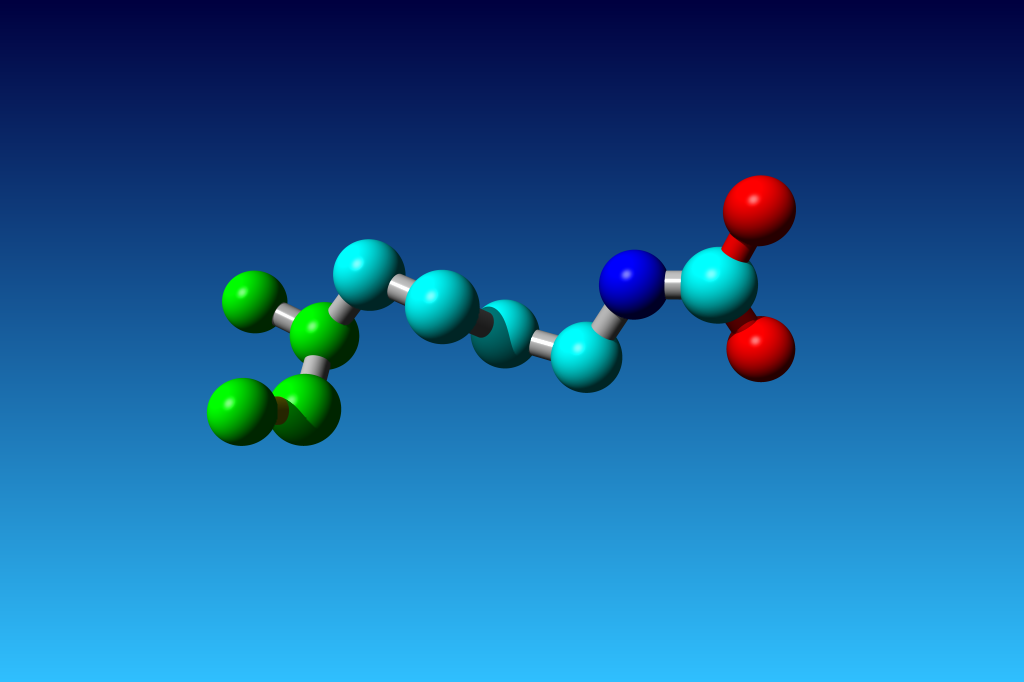
|
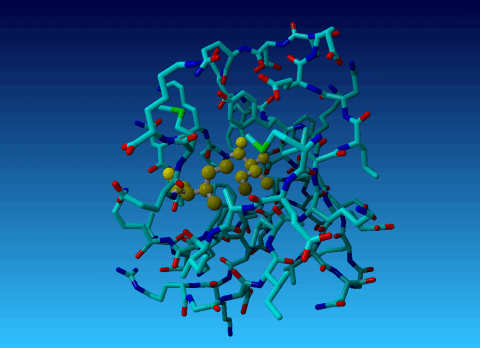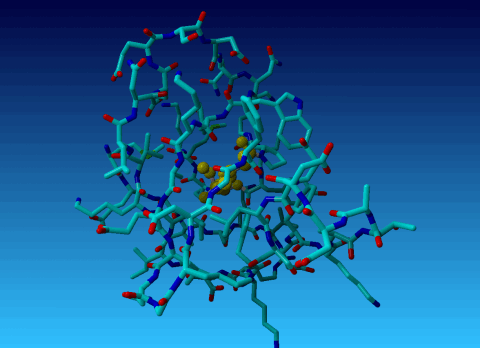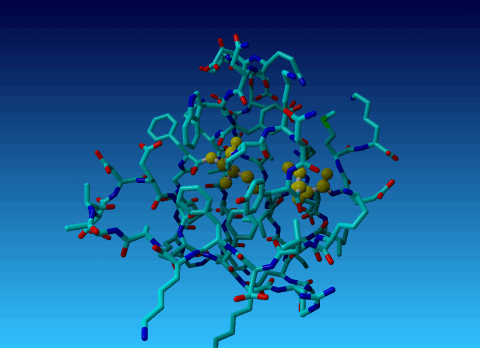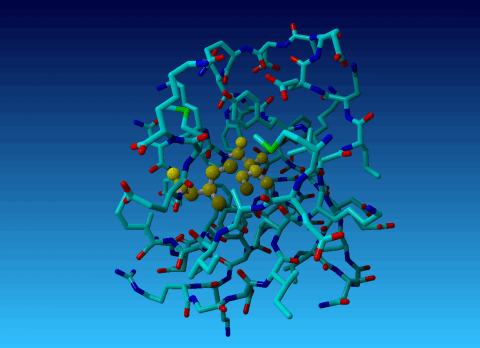
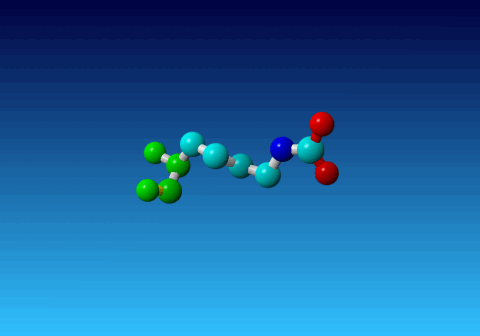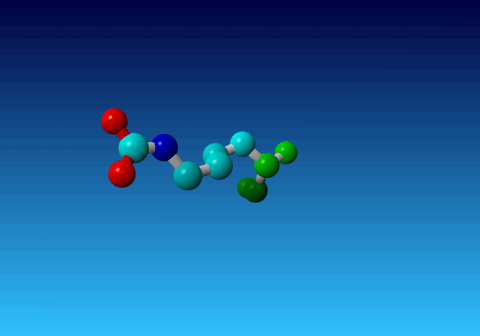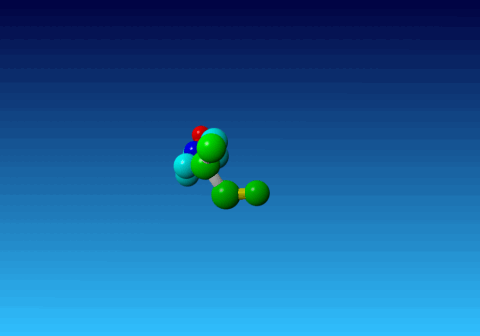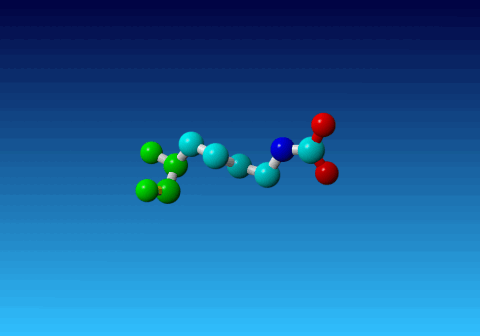
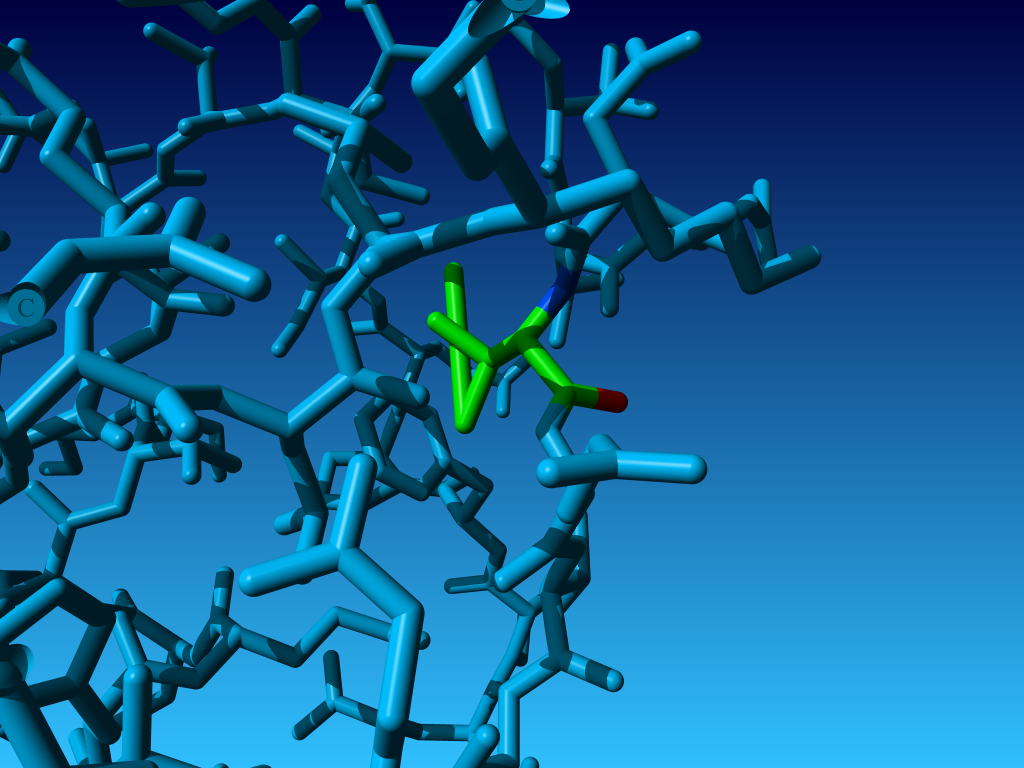
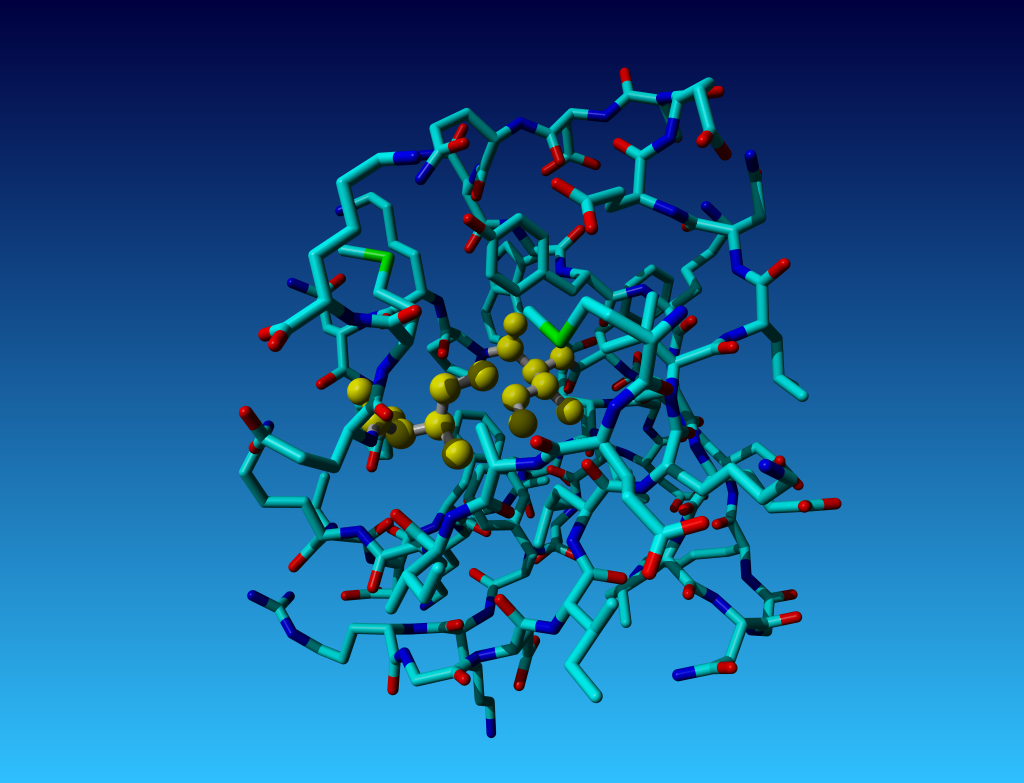
Figure 33. KCX (a NZ-carboxylated lysine), with its backbone atoms coloured
bright green. In this residue the 'official' atom names are: N CA C O
CB CG CD CE NZ CH OX1 OX2. The Greek alphabet rule thus is
disobeyed. I can imagine that one decided to skip θ as its European
equivalent has two characters (th), but the ξ now chosen instead seems a
rather illogical choice.
|