Articles
There is a large number of articles associated with the WHAT IF project.
A number of those describe technologies that we designed and/or implemented
especially for WHAT IF and/or WHAT_CHECK. This page lists a selection of
30 years of software development results.
WHAT IF
If you use WHAT IF, please refer to:
WHAT IF: A molecular modeling and drug design program.
G.Vriend, J. Mol. Graph. (1990) 8, 52-56.
YASARA Twinset
If you use the YASARA twinset, please refer to:
YASARA View - molecular graphics for all devices - from smartphones to workstations.
E.Krieger, G.Vriend, Bioinformatics (2014) 30(20), 2981-2982.
WHAT_CHECK
If you use WHAT_CHECK, please refer to:
Errors in protein structures.
R.W.W. Hooft, G. Vriend, C. Sander, E.E. Abola, Nature (1996) 381, 272-272.
Please be aware that we also have a special
WHAT_CHECK
page that includes extensive documentation about all options in the
WHAT IF servers
, and in the
PDBREPORT database
.
WHAT IF servers
If you used one of the WWW-based servers, please refer to:
Homology modeling, model and software evaluation: three related
resources.
R.Rodriguez, G.Chinea, N.Lopez, T,Pons, G.Vriend CABIOS (1998) 14, 523-528.
WHAT IF web services
Many WHAT IF options are available as web services. Please look at
the WSDL
or at the
Help
pages for these services. And if you use them, please cite:
WIWS: a protein structure bioinformatics Web service collection.
M.L.Hekkelman, T.A.Te Beek, S.R. Pettifer, D. Thorne, T.K. Attwood, G. Vriend
NAR (2010) 38, W719-723.
WHAT IF technologies
The WHAT IF project has been used to implement a series of ideas. This section
describes (briefly) some of these techniques.
The SEARCH menu
We all know how to work with protein structure databases. You can ask questions like
"give me all proteins that contain a proline in the middle of a helix". But what if
you want to know if there is a proline in a helix somewhere in the one protein
you are studying, and that accidentally is not available in the database?
In such a case you can use WHAT IF's
SEARCH menu. This menu
is described in the article:
Parameter relation rows: a query system for protein structure function
relationships.
G.Vriend, Prot. Engin. (1990) 4, 221-223.
The SCAN3D menu
The SCAN3D menu
allows you to do similar things as the SEARCH menu, but
SCAN3D runs over the internal database, and not over the SOUP.
A novel search method for protein sequence-structure relations,
using property profiles.
G. Vriend, C. Sander, P.F.W. Stouten, Prot. Engin. (1994) 7, 23-29.
We were later asked to write a short review about roughly this same topic:
How to extract non-trivial information from protein structure and
sequence data.
P. Stouten, G. Vriend. CDA News (1992) 7/5, 18-23.
The SUPPOS menu
One often wants to compare protein structures. To do so, it is very useful to
superpose them first.
This article describes a method that allows you to find the best
superpositioning of two protein structures. Many programs could do
this already in 1991, but this WHAT IF
option was the first algorithm that did not require any manual
pre-alignment. The main idea is that a distance geometry algorithm
is used to very quickly detect all fragments in the two proteins
similar that have similar backbone conformations. Clustering these
fragments gives the largest possible sub-structure between the two
proteins. Many people have written programs based on this
principle. Later, Liisa Holm has written the DALI method which, I think,
is the only method that works a bit better than our method.
The WHAT IF superposition method is described in
the SUPPOS menu of the writeup.
Unfortunately, the WHAT IF SUPPOS menu was to far ahead of its time.
Nowadays, one new structure superposition program is written
somewhere in the world every six months or so. And these young kid
authors don't read (and thus don't cite) articles from 1991...
Detection of common three-dimensional substructures in proteins
.
G. Vriend, C. Sander, PROTEINS (1991) 11, 52-58.
The WALPRF menu
WHAT IF has a series of sequence menus. One of them, the
WALPRF menu
is set up to do profile based sequence alignments. This menu was written,
originally, for this article (and to do the correlation analyses
for the next article down this list):
Modelling of transmembrane seven helix bundles.
P. Cronet, C. Sander, G. Vriend, Prot. Engin. (1993) 6, 59-64.
Everybody in the GPCR field has once tried to model GPCRs based on
bacteriorhodopsin. The article by Philipe Cronet is our attempt.
If you read it well, you will see that it is a rather smart threading
method (so, we were among the first or the first to do threading,
we just did not know how to call it at that moment...). Do not think that our
models are better than the others, they are equally wrong, but in
a different way. In this article we put more trust in experimental
data than in our ideas about what it should be theoretically, and I
think that that made this article rather unique among the 500
articles that around 1990, together with ours, poluted literature
with GPCR models.
The WALCOR menu
This article was the beginning of the real GPCR work, and the
first of our series of articles on correlated mutations. Although
that idea was not new, we introduced here the concept of
functional correlations. This work is described in the
WALIGN menu.
A model for G-protein coupled receptors.
L. Oliveira, A. Paiva, G. Vriend, J. Comp. Aided Mol. Des.
(1993) 7, 649-658.
The QUALTY menu
The QUALTY menu was
WHAT IF's first step on the long path
of protein structure validation.
This article describes the packing quality control module of
WHAT IF. This method is also called DACA for Directional Atomic
Contact Analysis. The idea is that the distribution of atom
types is determined around amino fragments. We assume that
the average distribution observed in the PDB is representative
for what can happen in nature. Unlike many other methods, we
do not spherically average the distribution around the fragment,
but we keep the x,y,z directionality of the contacts. The
quality of any structure is now easily determined by a
convolution of the average distributions and the observed
contacts in the protein to be checked.
Quality control of protein models: Directional atomic
contact analysis.
G. Vriend, C. Sander. J.Appl.Cryst. (1993) 26, 47-60.
The MUTATE etc menu
WHAT IF has been used extensively to predict point mutations
in proteins. The idea (free after Jones and Thirup) is to
extract from the database fragments that fit well on the fragment
that has the residue to be mutated in the middle. If the fragments
are selected to only have the desired (novel) residue in the middle
one obtains a so-called position specific specific rotamers.
These next three siles, extracted from an old homology
modelling seminar, illustrate that:
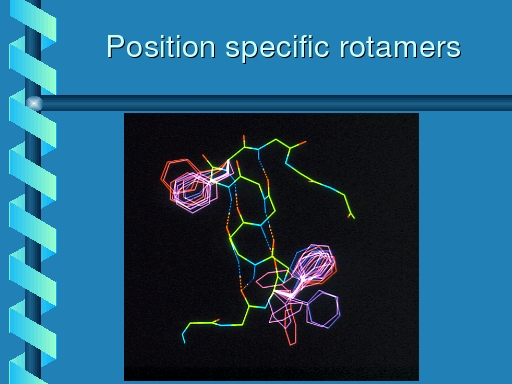
|
Rotamer distribution for phenylalanine at two positions in
a protein. The one position clearly has a preference for
one of the three primary rotamers; at the other position there
also seems to be one favourite, but the preference is slightly
less strong.
|
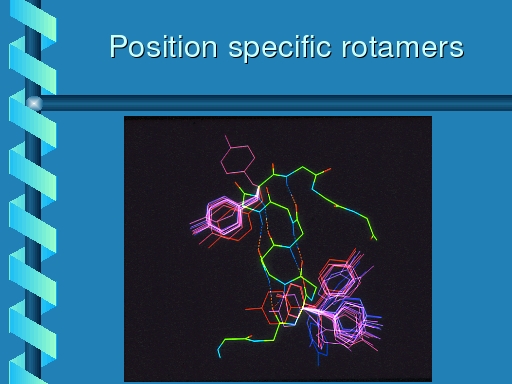
|
Rotamer distributions for tyrosine at the same two positions.
The top left rotamer shows the same distribution as observed for
phenylalanine, but the bottom right one is much more bimodal.
|
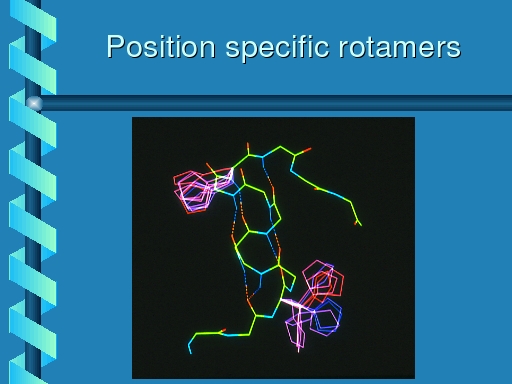
|
Rotamer distributions for histidine at the same two positions.
The top left rotamer shows the same distribution as observed for
phenylalanine, and tyrosine, but the botton right one now is equally
populated in each of its three primary rotamers.
The DGLOOP menu in WHAT IF holds a large
number of rotamer related options.
|
This work was published in:
The use of position specific rotamers in model building by
homology.
G. Chinea, G. Padron, R.W.W.Hooft, C.Sander, G.Vriend,
PROTEINS (1995) 23, 415-421.
WHAT_CHECK (is WHAT IF's CHECK menu)
All protein structures solved by NMR or X-ray (that have been
deposited in the PDB) contain errors ranging from small problems
like bond lengths that are a bit too long till catastrophical
things like backwards threaded sequences.
Rob Hooft wrote most options of the
CHECK menu in WHAT IF. The first
thing published was the software to reconstruct the full cell from
the cell and scale information provided in the PDB file. It is
amazing how many different errors the crystallographers can make
in so few information:
Reconstruction of symmetry related molecules from protein data
bank (PDB) files.
R. Hooft, C. Sander, G. Vriend, J. Appl. Cryst. (1994) 27,
1006-1009.
We have done a series of callibrations on the data underlying the
validation options. Like:
On the complexity of Engh and Huber refinement restraints: the
angle τ as example.
W. Touw, G. Vriend. Acta Crystallogr D (2010) 66 1341-1350.
An important aspect of the CHECK facility is the option to get
all hydrogenbonds right. More than 10% of all asparagines,
glutamines, and histidines in the PDB are flipped 180 degrees
about their chi-2, chi-3, and chi-2 side chain torsion angles,
respectively. The hydrogenbond optimization software finds these
problems:
Positioning hydrogen atoms by optimizing hydrogen-bond
networks in protein structures.
R.W.W.Hooft, C.Sander, G.Vriend, PROTEINS (1996) 26, 363-376.
Side chain planarity has been close to random in the early
days of X-ray refinement and even today there are still authors
of well-known refinement packages (e.g. D.T. and A.B.) who
think they know better than the results that can be
extracted from a study of the CSD. Anyway, structures refined
with those softwares are invariably flagged by:
Verification of protein structures: Side-chain planarity.
R.W.W. Hooft, C.Sander and G.Vriend,
J. Appl. Cryst. (1996) 29, 714-716.
This paper describes the verification of side-chain planarity
by WHAT IF and WHAT_CHECK. It also describes the construction of
a representative list of PDB files as used in the WHAT IF database.
This latter facility is available as the
PDBSELECT
database.
The Ramachandran plot is probably the best determinant of protein
quality. This was first realized by Thornthon and Laskowsky. Others
(e.g. T.A.J.) later tried to improve their PROCHECK software by
tightening the borders a bit. Rob's approach to do things with
proper statistics and with residue specificity, however, was a
significant improvement:
Objectively judging the quality of a protein structure
from a Ramachandran plot.
R.W.W. Hooft, C.Sander and G.Vriend, CABIOS (1997), 13, 425-430.
The X-ray cell dimension validation of WHAT IF is decribed in:
Some WHAT_CHECK checks explained.
R.W.W.Hooft, G.Vriend, PDB Newsletter. (1998) April volume.
The quality of the quality checks was explained in:
Who checks the checkers? Four validation tools applied to
eight atomic resolution structures.
K.Wilson, C.Sander, R.W.W.Hooft, G.Vriend, et al.
J.Mol.Biol. (1998) 276,417-436.
Validating protein structures (and models) is one of our
hobbies. This article (which has 20 authors, the whole validation
consortium is on it...) describes the results of validating some
structures solved at atomic resolution (around 1.0 A) that thus
were supposed to be guaranteed correct. This study revealed some
problems in the ultra highly refined structures and some problems
in the refinement programs. The general conclusion should be that
the validation programs are actually working very well, and that
remarks by some crystallographers that "these validation programs
give very many false positives" really are not supported by
experimental verification.
Rob Hooft mainly wrote X-ray specific validation options, and he wrote
the WHAT IF infra-structure for validation in general. Other
people have worked on NMR structure validation. Jurgen Doreleijers
has for many years worked on getting NMR ensembles simply
administratively correct. A mamoth task...
Validation of NMR structures of proteins and nucleic acids:
hydrogen geometry and nomenclature.
J.F.Dorelijers, G.Vriend, M.L.Raves, R.Kaptein
Proteins (1999) 37, 404-416.
Molecular dynamics
The Berendsen group in Groningen pioneerd the concept of 'essential
dynamics' (Amadei A, Linssen AB, Berendsen HJ. Essential dynamics of
proteins. Proteins. 1993 Dec;17(4):412-25.). Initially, most essential
dynamics software was implemented in WHAT IF. This led to a series of
articles. One of the older ones is:
The essential dynamics of thermolysin: confirmation of the
hinge-bending motions and comparison of simulations in vaccuum and water.
D. van Aalten, A. Amadei, A. Linssen, V. Eijsink, G. Vriend and H.
Berendsen PROTEINS (1995) 22,45-54.
Nowadays most essential dynamics software is incorporated in GROMACS,
and in version 6.1 WHAT IF will interface to this new essential dynamics
software again.
Ligand interpretation with PRODRUG
WHAT IF uses PRODRUG to interpret ligands:
PRODRG: a program for generating molecular topologies and unique
molecular descriptors from coordinates of small molecules.
D.M.F.v.Aalten, R.Bywater, J.B.C.Findlay, M.Hendlich, R.W.W.Hooft,
G.Vriend JCAMD (1996) 10, 255-262.
This paper describes a program that can be used to get WHAT IF
topology entries for small molecules. The program can be used by
everybody via a server. Additionally, a MOL2 file and a GROMOS
topology are generated.
This software is not associated with any menu. It is simply
integrated in the PDB-file interpretation option in WHAT IF.
Protein mobility and flexibility
This is Bert de Groot's domain. He is the main author
of the CONCOORD software that can be used to predict
the outcome of MD simulations:
Prediction of Protein Conformational Freedom from Distance
Constraints.
B.L.de Groot, D.M.F.van Aalten, R.M.Scheek, A.Amadei,
G.Vriend, H.J.C.Berendsen PROTEINS (1997) 29,240-251.
Electrostatics and pKa
One day, in the past, WHAT IF could deal with an improved method for
calculation of electrostatics and pKa values:
Improving macromolecular electrostatics calculations.
J.E. Nielsen, K.V. Andersen, B. Honig, R.W. Hooft, G. Klebe,
G. Vriend, R.C. Wade
Prot.Eng. (1999) 12(8), 657-662.
Good electrostatics is only the first step towards
the more lofty goal of pKa calculations:
Optimizing the Hydrogen-Bond Network in Poisson-Boltzmann
Equation-based pKa Calculations.
Nielsen, JE., Vriend G. Proteins 2001 43: 403-412.
Unfortunately, Jens no longer could find the time to maintain the electrostatics menu in WHAT IF.
So, if you need these calculations, contact him directly.
Graphics
WHAT IF was for a long time the only software that could
display molecules with full 3D stereo using stereo glasses
from the computer-game industrie, rather that the horrifically
expensive SGI glasses that only worked on expensive
workstations:
Full window stereo
Rodriguez R, Chinea G, Lopez N, Vriend G
J Mol Graph & Mod (1999) 17, 310-313.
Visualisation is the bioinformaticist's most important tool
for the study of macromolecules, and being able to see molecules
in stereo is a crucial aspect. Stereo vision is based on the
principle that each eye is presented with the best possible
image of what it would have seen if the object was really
there in 3D. The simplest approach to stereo vision is to
display the right eye picture on the right half of the screen
and the left eye picture on the left half while using a mirror
system to ensure that each eye sees what it is supposed to see.
More expensive workstations use hardware to alternatively
display the left and right eye pictures while synchronously
blocking the transparency in the right or left lens of the
special glasses worn by the user. We present here some simple
software that uses inexpensive hardware, originally designed
for the computer game industry, to make full screen stereo
available on Linux-based PCs. The quality of the stereo
vision is similar to the top-of-the -line graphics workstations
that are capable of quad-buffering. This stereo option has
been incorporated in the X11 based version of WHAT IF, but
the stereo source code is freely available and can easily be
incorporated in other visualization packages.