Material linked from bioinformatics course
While many scientific disciplines face huge difficulties when trying
to experimentally validate theoretical predictions, protein modelling
is in a fortunate situation: since 1994, the biennial CASP ('Critical
Assessment of Structure Prediction') contests (Moult et al. 2007)
provide an ideal opportunity to evaluate the accuracy of today's
many protein structure prediction methods. During each CASP
season (lasting about four months once every two years), about 200
research groups try to predict the structures of ~100 proteins, the
CASP targets. The target sequences are provided to CASP by structural
biology labs just before the corresponding structures are solved. The
predictions are thus true blind predictions, allowing to measure
performance in realistic test cases, locating areas of progress as
well as yet unsolved problems.
CASP regularly shows that the eight homology modelling steps summarized
here allow in many cases building reliable models, from which a lot of
structural and functional insights can be derived. However, these eight
steps are unfortunately not sufficient to actually solve the protein
structure prediction problem via homology modelling as soon as enough
templates become available.
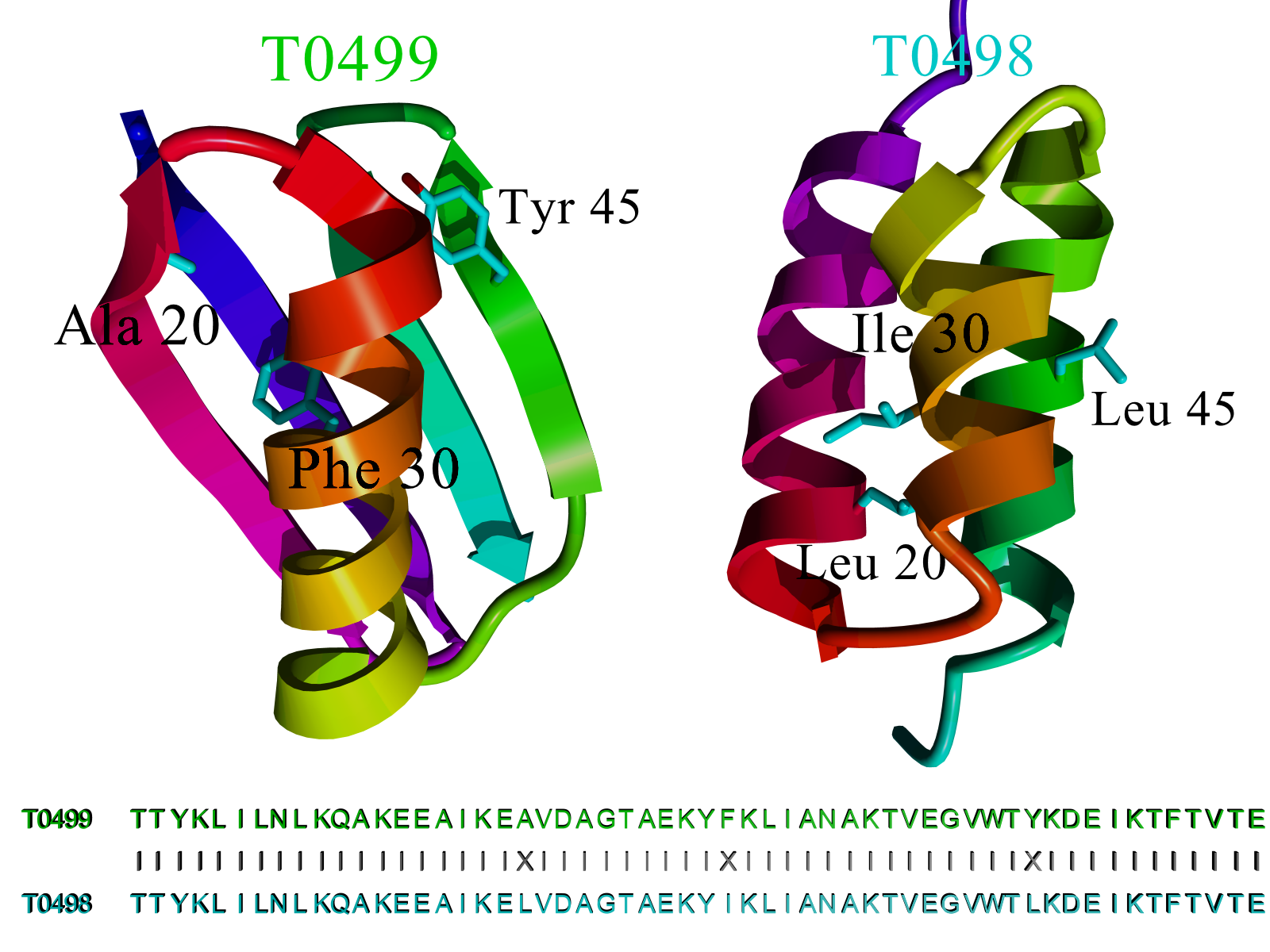
|
Figure 26. Comparison of CASP8 targets T0498 and T0499. The sequences of both
proteins are 95% (53 of 56) identical (only residues 20, 30 and 45
differ), yet the structures are totally different. Classic homology
modelling predicted T0499 correctly (which looks like the related
homology modelling templates in the PDB), but failed completely
for T0498. Since the structures of T0498 and T0499 have not been
released yet, this figure is based on a closely related pair
with PDB IDs 2jws and 2jwu from the same authors, who showed
by NMR spectroscopy that these two structures look essentially
the same as T0498 and T0499 (He et al. 2008).
|
The figure shows CASP8 targets T0498 and T0499: both proteins
are 56 amino acids long, 53 of which are conserved (95% sequence
identity). Still, the two structures are entirely different; just
three point mutations completely change the fold. While this is an
extreme example of human protein engineering art (He et al. 2008),
also naturally occurring proteins with similar sequences often
show surprising structural diversity (Kosloff and Kolodny 2008),
letting classic homology modelling fail miserably. The prion
protein (Prusiner 1998) and other amyloid-forming proteins
provide an even more dramatic case; here 100% identical sequences
can exist in two totally different structures. Obviously, the
homology modelling problem is tightly intertwined with the more
general protein folding problem itself. Even if a close template
is available, there can always be structurally diverging regions,
which are either expected from the poor local alignment, or
unexpectedly caused by critical point mutations, or widely differing
crystal packing contacts.
The only way to handle these difficult cases is to apply more general
ab initio folding algorithms, which do not depend on template structures,
but try to simulate the complete folding process from the stretched-out
conformation. As it turns out, this 'one-algorithm-for-everything'
approach is the currently most successful one at CASP (Chivian et
al. 2003; Pandit et al. 2006): if available, it uses known
templates (or fragments thereof) only to guide the search, but
does not depend on them. As a side-effect, this allows to build
hybrid-models, combining the best parts from multiple templates.
Despite these encouraging developments, the protein folding problem
is far from solved. The best models are still built by those who got
the alignment right in the first place, which unfortunately implies
that structural diversity is often missed: one cannot yet ignore the
difficult-to-align regions and simply predict them with ab initio
folding instead. The sequence alignment problem thus remains an
active research field for years to come.
Noteworthy progress has been made with model optimization to
bridge the structural gap between initial model and target. While
in the early days of CASP, predictors were well advised to keep the
backbone of their model fixed (the 'frozen core approach'), simply
because the danger of messing up the model was just too large, the
situation is quite different today: force field accuracy (Krieger
et al. 2004) and sampling efficiency (Misura et al. 2006) have
improved to a level that allows well performing methods like
Modeller-CSA (Joo et al. 2008), Rosetta (Chivian et al. 2003),
undertaker (Vriend 1990), and YASARA (Krieger et al. 2009) to
free all atoms during the refinement, often moving models
considerably closer to the target.
While homology modelling currently focuses on the protein in
a model, other entities, i.e. carbohydrates, small molecules,
and ions, also make up important parts of certain proteins and
protein complexes. For instance, zinc atoms in so-called zinc
fingers are important for the stability of the protein, and a
common protein like haemoglobin would be useless without its
haem groups and the iron atoms therein. Carbohydrates in
glycoproteins perform numerous functions, ranging from
providing stability to signalling and labelling for intra-cellular
transport (Lütteke 2009). The many roles of non-protein entities
make it obvious that homology modelling should look beyond the
protein. A complete model should thus be more than a 3D representation
of an amino acid sequence. One major challenge for homology modelling
is recognizing binding sites for non-protein entities.
Drug docking software (e.g. Rary et al. 1996; Nabuurs et al. 2007)
can be used to detect the binding sites of compounds such as heme
groups or co-enzymes. However, relevant biological information is
needed to select compounds that may be bound to the protein. Copying
the binding site from the template structure is the simplest method,
but does not work for ab initio folding models. For such models,
spectroscopic analysis of the protein can provide insight on which
compounds are bound. This approach is not limited to homology
modelling; X-ray crystallography can also benefit from spectroscopic
analysis of a protein to identify a bound compound (Chen et al. 2002).
Incorporating ions can be an additional step of the modelling process.
Nayal and Di Cera (1996) have suggested a method to detect sodium binding
sites in protein structures which can be extended to detect various
other ion binding sites. Of course, any additional experimental data
can guide this ion site detection process. Especially tightly bound
functional ions that co-purify with the protein can be detected by
means of spectroscopic analysis. A significant number of PDB files
have bound ions or water molecules that were erroneously assigned.
We have observed , and Na+, K+, Mg2+, Ca2+, and NH4+ ions that
should actually be one of the others in this list. This is the
result of X-ray spectroscopy having difficulties distinguishing
between H2O, NH4+, Na+ and Mg2+ because these entities have
equally many electrons, and so do K+ and Ca2+.
The power of force-field based model optimization methods can be
reduced significantly when such problems include a difference in
the ionic charge. It is therefore very important to (experimentally)
validate the ions in template structures when these are important
for the final homology model.
Carbohydrates can be modelled at the final stage of the homology
modelling process, but this does not always reflect the protein
folding process. Carbohydrates are not only added in post-translational
modification, but also
during the protein expression by the ribosome. They are important
in the protein folding process and the detection of misfolded
proteins (Parodi 2002). It may therefore prove interesting to
add the necessary carbohydrates to the unfolded protein before
ab initio folding. Apart from their role in protein folding,
carbohydrates are sometimes important in oligomerisation of
proteins. For instance, the neuraminidase protein from influenza
shows different glycosylation states in its monomeric, dimeric,
and tetrameric states. The carbohydrates in tetrameric state
provide extra stability and, in the case of the
Spanish flu influenza virus, resistance to trypsin digestion
leading to increased virulence (Wu et al. 2009).
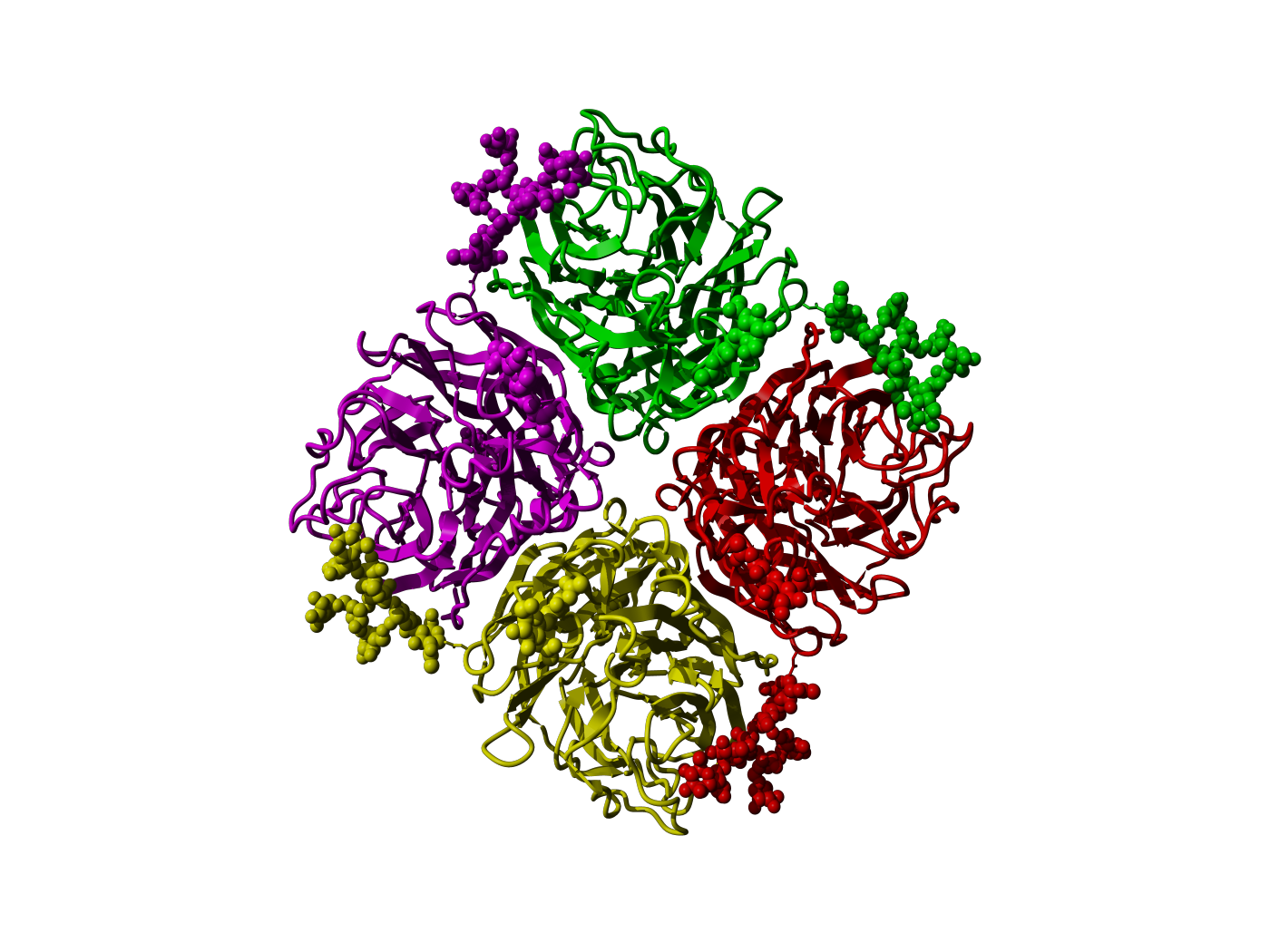
|
Figure 27. Tetrameric form of whale influenza neuraminidase (PDBid 2r8h,
Smith et al. 2006), coloured by monomer. Protein chains are
displayed in ribbon representation, carbohydrate atoms in ball
representation. The carbohydrates of one monomer interact with
the adjacent monomer thus stabilizing the tetramer.
|
This shows the vital (and sometimes lethal) importance of
considering carbohydrates in homology models.