GPCR activation
What moves where?
Introduction
GPCRs
Structure
Activation
The rhodopsin model
Ligand binding GPCRs
Approach
Random Forest
Variable Importance
GPCRs

G protein-coupled receptors (GPCRs) are responsible for the majority of cellular responses to external signals. They represent the largest and arguably the most diverse superfamily of membrane receptors represented in every eukaryotic cell. Over a 1000 human genes encode GPCRs. The ligands that bind or otherwise activate these receptors are heterogeneous and include photons, odours, pheromones, hormones, ions, neurotransmitters and proteases. GPCRs transmit signals from outside the cell to amplification cascades controlling sight, taste, smell, slow neurotransmission, cell division, etc.62
At first, in 1994, Attwood and Findlay categorized the superfamily into six classes (A–F) based on sequence homology and functional similarity. Later, our comprehensive phylogenetic analysis of the human repertoire provided the GRAFS classification. This system grouped the mammalian GPCR repertoire into five main families; Rhodopsin (Class A), Adhesion (Class B), Glutamate (Class C), Frizzled (Class F), and Secretin (Class B).6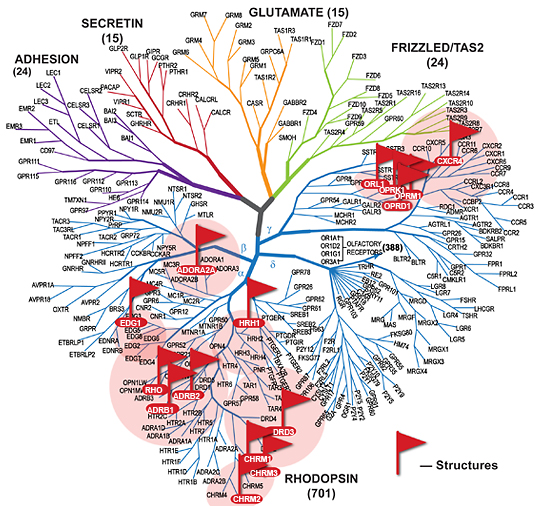 The Rhodopsin family is the largest with 683 members in humans 7, characterized by short N-termini and interactions with a broad variety of ligands.
The Glutamate family is distinguished with long N-termini which act as the endogenous ligand binding region.
The Adhesion receptors have long N-termini which contain a plethora of multiple domains while the Frizzled receptors have long cysteine-rich N-termini 8.
The Rhodopsin family is the largest with 683 members in humans 7, characterized by short N-termini and interactions with a broad variety of ligands.
The Glutamate family is distinguished with long N-termini which act as the endogenous ligand binding region.
The Adhesion receptors have long N-termini which contain a plethora of multiple domains while the Frizzled receptors have long cysteine-rich N-termini 8.
This scala of different GPCRs has a scala of different G protein they can bind. Subsequent to initial cloning efforts, cloning by homology has defined the human G-proteins to derive from 35 genes, 16 encoding alpha-subunits, five beta and 14 gamma. All function as guanine nucleotide exchange on-off switches and are mechanistically similar to other proteins that are enzymatic GTPases.9 Heterotrimeric G proteins can be broadly categorized into four major classes based on the identity of the β subunit: Gs, Gi/o, Gq/11, and G12/13.11
Structure
To understand the function of GPCRs at the molecular level, it is fundamental to investigate the nature of the structural rearrangements that couple ligand binding to receptor-dependent activation of downstream signalling pathways. All GPCRs contain a bundle of seven helices spanning the membrane (7TM) connected by three intracellular (ICL) and three extracellular loops (ECL).
Rhodopsin was the first GPCR whose crystal structure was determined to high resolution 35. Since then a number of structures were elucidated by crystallography.
Recently more and more class A GPCR structures besides Rhodopsin became available. All showing a highly resembling structure.
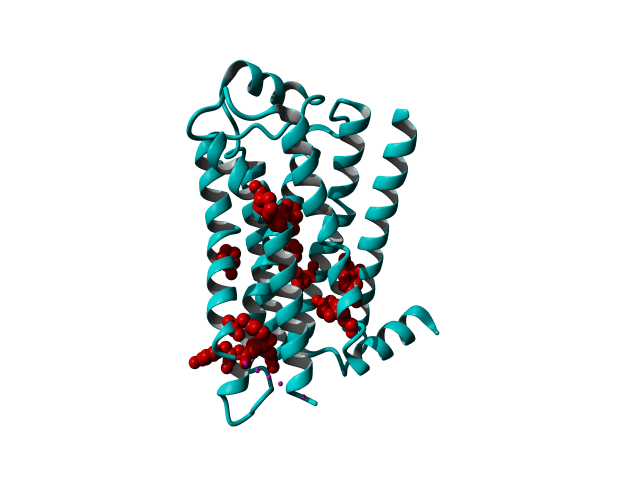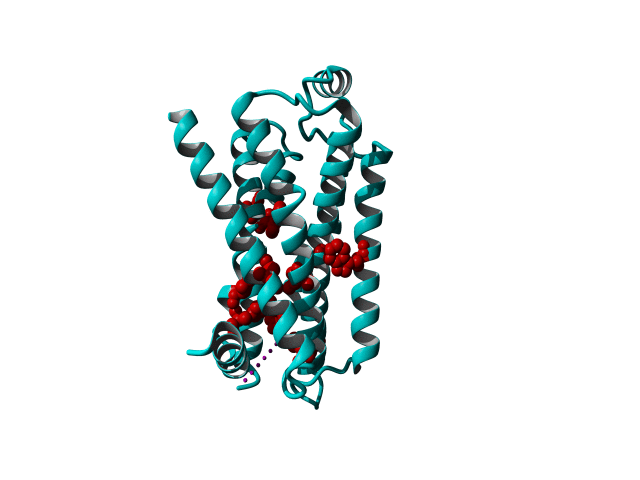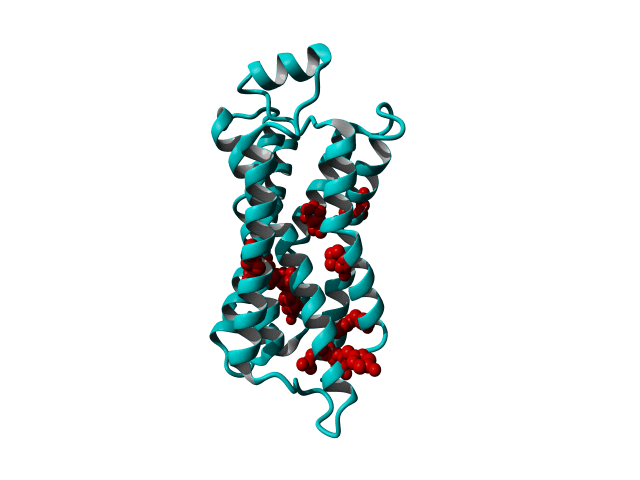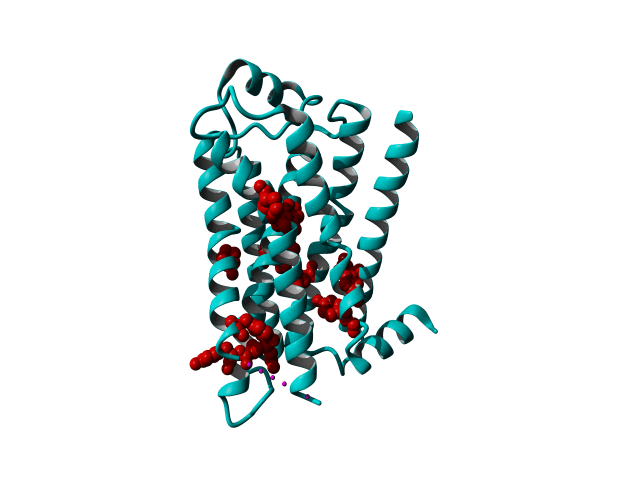 GPCRs are very flexible proteins and are located in the fatty environment of the cell membrane.
To allow these flexible proteins to assume a rigid structure, which is necessary in a crystal structure, they were modified to great extent.
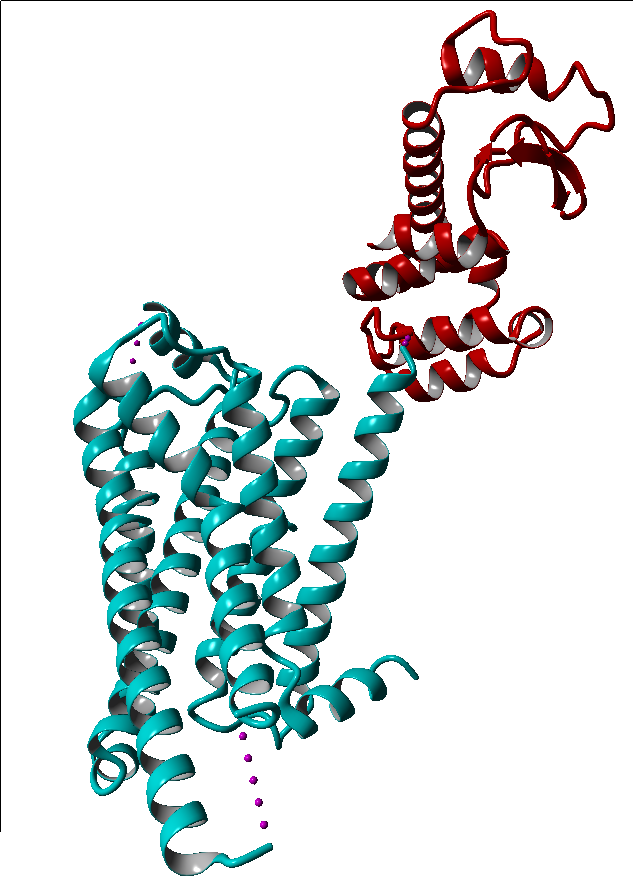
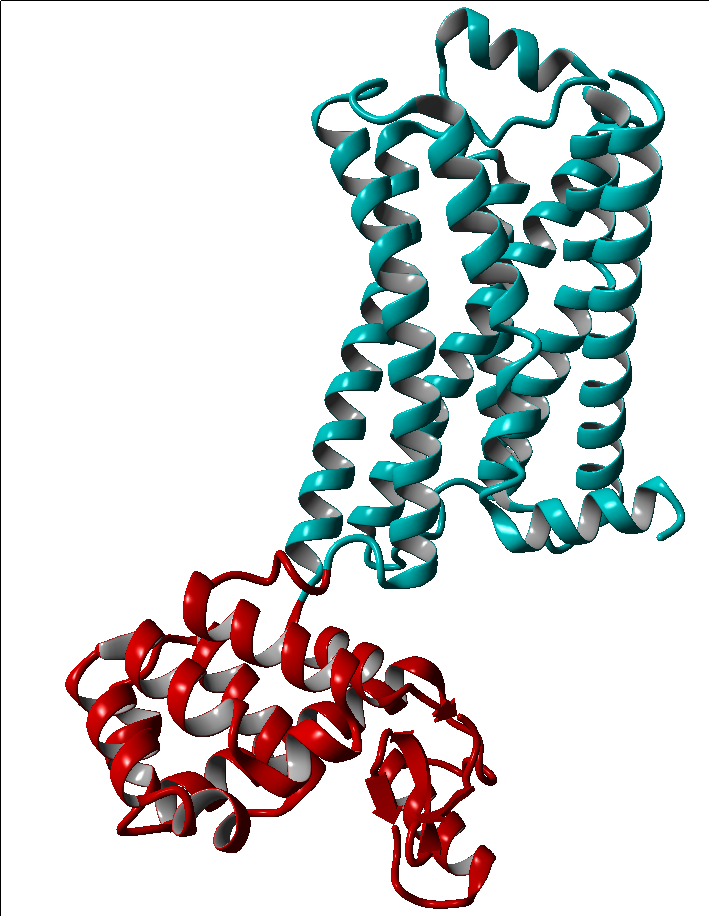
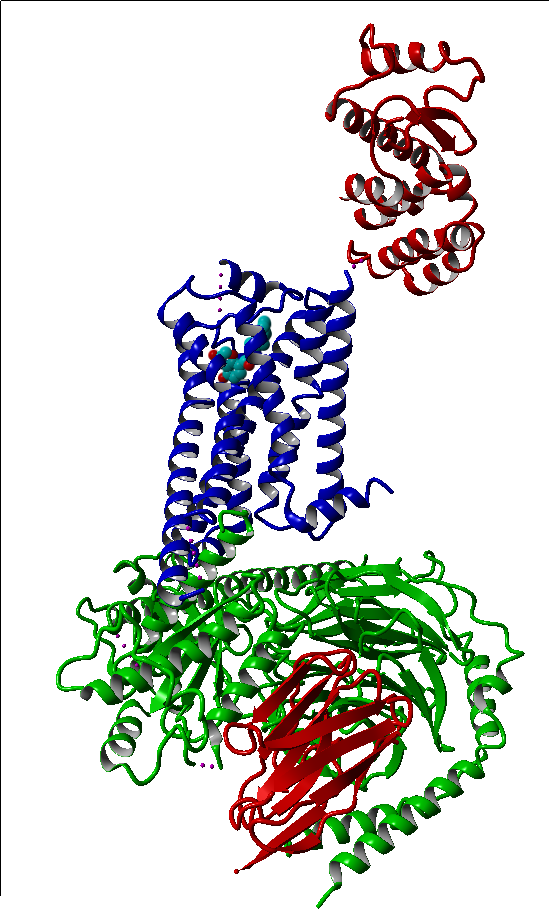
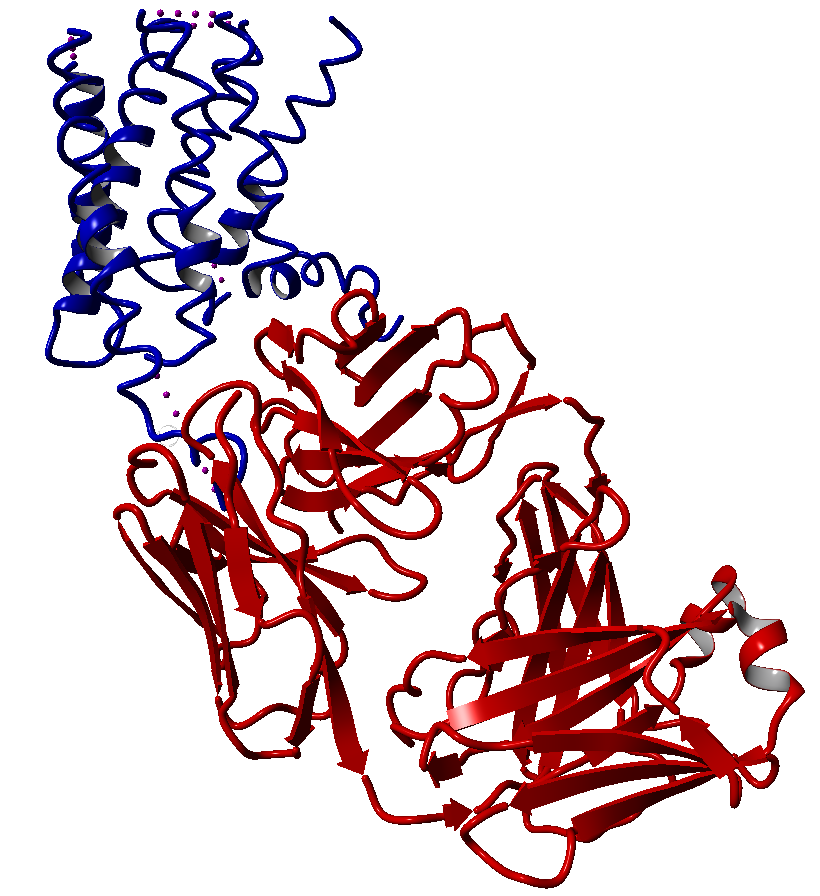
These modifications include thermostabilizing mutations, T4 Lysozyme fusion (Fig. 1, 2, Anti- or Nano-body binding or even a combination of several (Fig).
Among Class A GPCR there are some residues that are highly conserved. In each transmembrane helix there is at least one residue that is highly conserved.
In the figure above you can see the most conserved residues shown in red. In TM1 the Asn at position 130 (Oliveira numbering) is highly conserved. In TM2 it's L220. In TM3 there is a highly conserved motif consisting of E/D339, R340 and Y341 the so-called E/DRY motif. In TM4 it's W420. In TM5 P520 and Y528 are highly conserved.
In TM6 C617, W618 and P620 forming the CWxP motif and in TM7 there's a motif called the NPxxY motif consisting of N729, P730 and Y733. There are many theories about the roles of these residues in the transduction of the signal between the ligand binding pocket and the G protein binding pocket.
GPCRs are very flexible proteins and are located in the fatty environment of the cell membrane.
To allow these flexible proteins to assume a rigid structure, which is necessary in a crystal structure, they were modified to great extent.
These modifications include thermostabilizing mutations, T4 Lysozyme fusion (Fig. 1, 2, Anti- or Nano-body binding or even a combination of several (Fig).
Among Class A GPCR there are some residues that are highly conserved. In each transmembrane helix there is at least one residue that is highly conserved.
In the figure above you can see the most conserved residues shown in red. In TM1 the Asn at position 130 (Oliveira numbering) is highly conserved. In TM2 it's L220. In TM3 there is a highly conserved motif consisting of E/D339, R340 and Y341 the so-called E/DRY motif. In TM4 it's W420. In TM5 P520 and Y528 are highly conserved.
In TM6 C617, W618 and P620 forming the CWxP motif and in TM7 there's a motif called the NPxxY motif consisting of N729, P730 and Y733. There are many theories about the roles of these residues in the transduction of the signal between the ligand binding pocket and the G protein binding pocket.
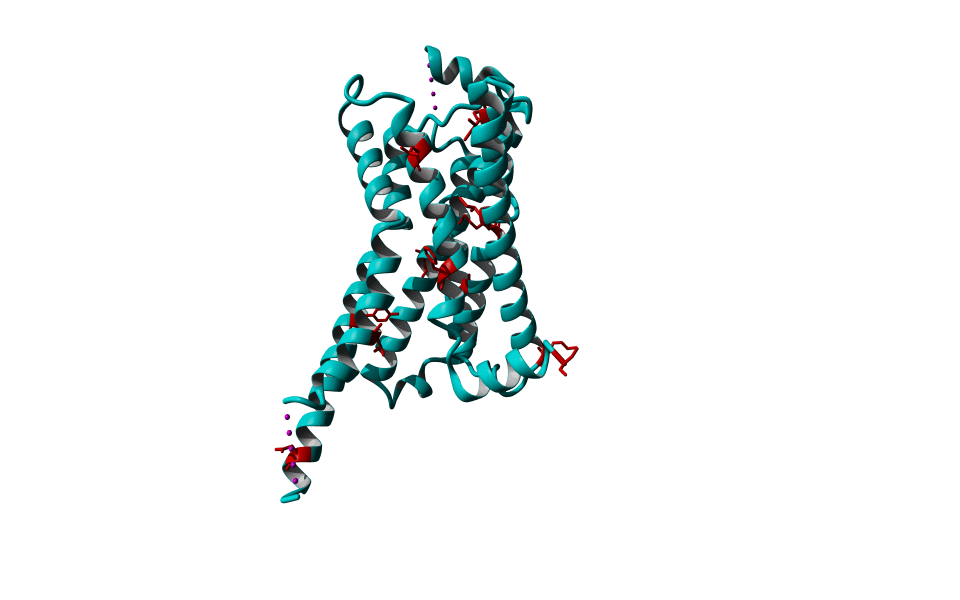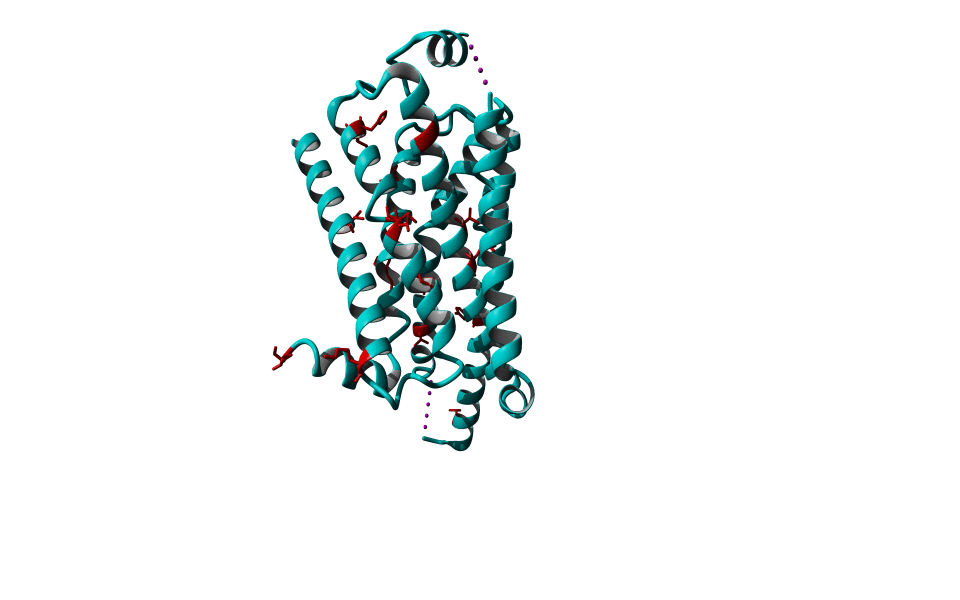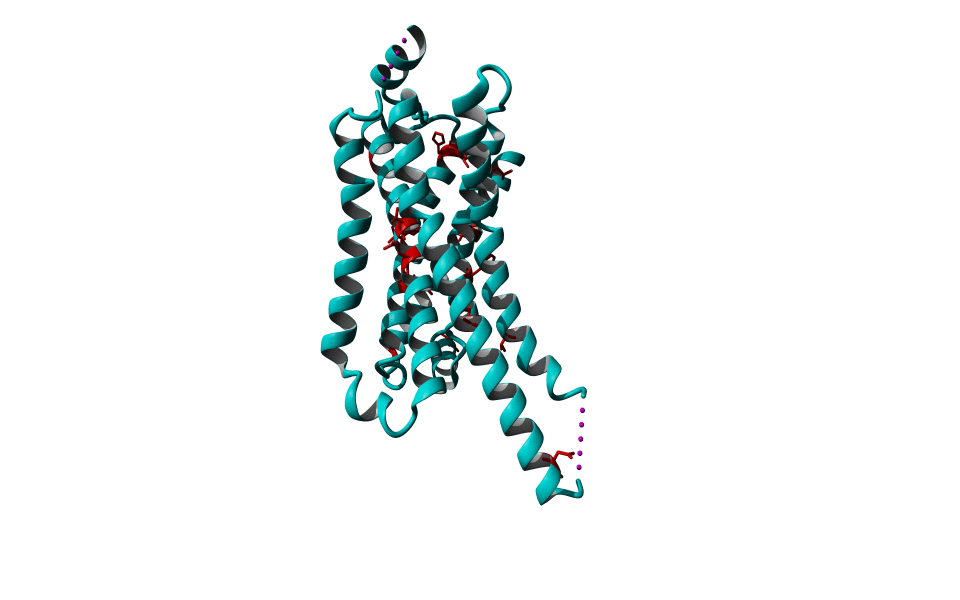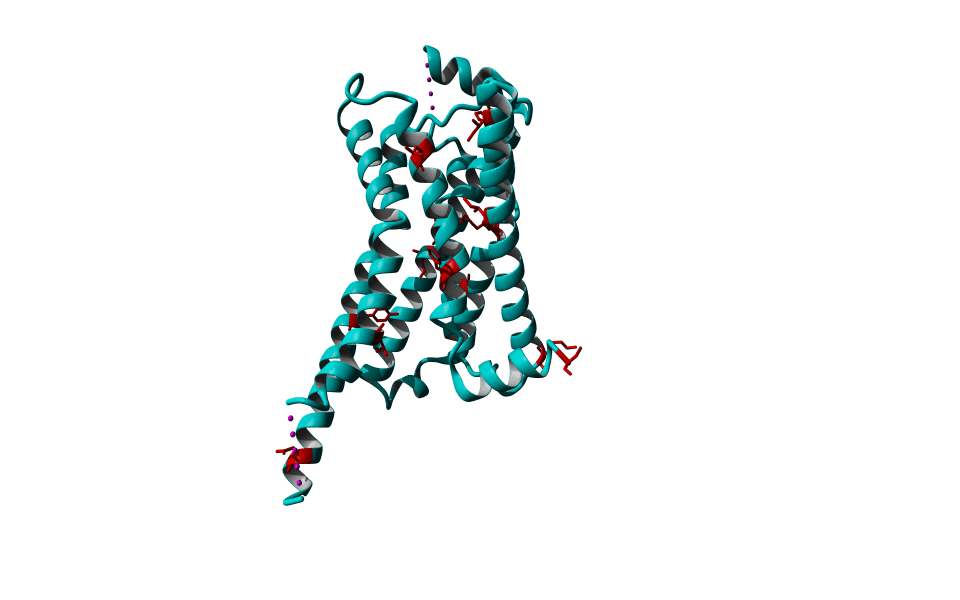
Activation
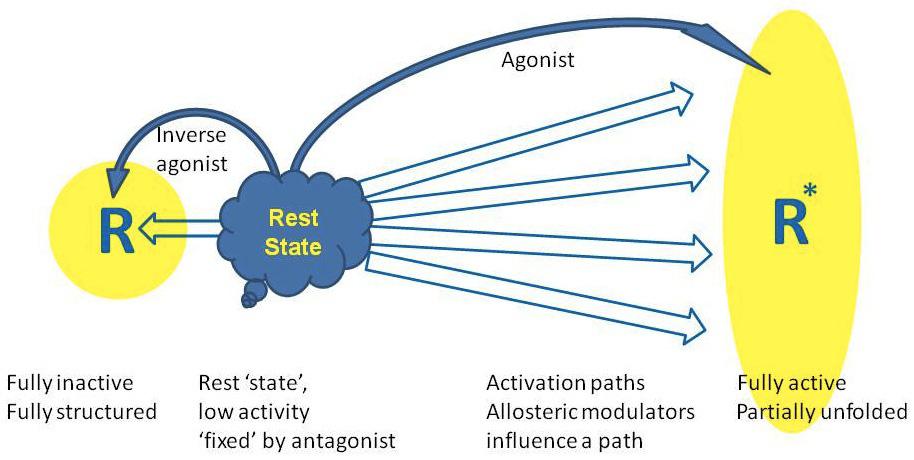
The repertoire of signalling activities by GPCRs is considerably more complex than envisioned in the early periods of research that led to the formulation of classical models (Fig). GPCR signalling can no longer be viewed as a single pathway consisting of a linear sequence of events.10
For a long time GPCRs were thought to perform a relatively straightforward role, namely coupling the binding of agonists to the activation of G proteins,
which in turn leads to modulation of other downstream effector proteins.
However, in recent years it has become clear that many GPCRs have much more complex signalling characteristics.
 Many GPCRs are constitutively active, and this allows for a fine-grained control of the amount of G-protein activation, being subject to regulation by agonist as well as inverse agonists 1.
It is now clear that G proteins exhibit some promiscuity and a single GPCR can couple to and signal via G proteins from multiple classes, which results in the propagation of signals through multiple biochemical pathways to achieve different cellular responses.12
Many GPCRs are constitutively active, and this allows for a fine-grained control of the amount of G-protein activation, being subject to regulation by agonist as well as inverse agonists 1.
It is now clear that G proteins exhibit some promiscuity and a single GPCR can couple to and signal via G proteins from multiple classes, which results in the propagation of signals through multiple biochemical pathways to achieve different cellular responses.12
 Also in the classical view, GPCRs signal exclusively via heterotrimeric G proteins to generate the cellular response. It is now apparent that G protein-independent signalling can occur within a cell via arrestin molecules to increase the diversity of cellular responses a single GPCR can generate. Arrestins in the classical view serve as proteins that bind phosphorylated GPCRs deactivate and, in most instances, internalize the receptor. The role arrestins play in the cell has now expanded to also include signalling functions. GPCRs can signal independently of G proteins via arrestins to mitogen-activated protein (MAP) kinases, which can regulate chemotaxis, apoptosis, cancer metastasis, and protein translation. Because of the capability of receptors to signal independently of the G protein, GPCRs are now sometimes referred to as seven-transmembrane receptors.2,13
Desensitization processes can involve multiple pathways, including phosphorylation events, arrestin-mediated receptor internalization, receptor recycling, and lysosomal degradation 3-5.
The reason for this multifaceted behaviour may be the fact that, while there are only about 1000 GPCRs that can be activated by an even smaller number of endogenous agonists, these receptors need to cater for many 1000s of different messages that the whole organism needs to be able to transmit internally. They can also form homo- and heterodimers, which cooperatively modulate signaling. For more information on dimerization, click here.
Also in the classical view, GPCRs signal exclusively via heterotrimeric G proteins to generate the cellular response. It is now apparent that G protein-independent signalling can occur within a cell via arrestin molecules to increase the diversity of cellular responses a single GPCR can generate. Arrestins in the classical view serve as proteins that bind phosphorylated GPCRs deactivate and, in most instances, internalize the receptor. The role arrestins play in the cell has now expanded to also include signalling functions. GPCRs can signal independently of G proteins via arrestins to mitogen-activated protein (MAP) kinases, which can regulate chemotaxis, apoptosis, cancer metastasis, and protein translation. Because of the capability of receptors to signal independently of the G protein, GPCRs are now sometimes referred to as seven-transmembrane receptors.2,13
Desensitization processes can involve multiple pathways, including phosphorylation events, arrestin-mediated receptor internalization, receptor recycling, and lysosomal degradation 3-5.
The reason for this multifaceted behaviour may be the fact that, while there are only about 1000 GPCRs that can be activated by an even smaller number of endogenous agonists, these receptors need to cater for many 1000s of different messages that the whole organism needs to be able to transmit internally. They can also form homo- and heterodimers, which cooperatively modulate signaling. For more information on dimerization, click here.
The rhodopsin model
One member of the visual receptor subfamily of GPCRs, rhodopsin, is by far the best structurally defined GPCR.
Rhodopsin has been studied extensively and it's structure has been used as a template for homology modelling, but rhodopsin is actually a special case among the whole GPCR family, because it has it's ligand covalently bound to Lys723 by a protonated Schiff base.
All visual receptors from humans to squid have the 11-cis isomer of retinal bound within the 7TM bundle. In pharmacological terms, the 11-cis retinal chromophore acts as an inverse agonist, when bound it reduces the basal activity of the apoprotein opsin.
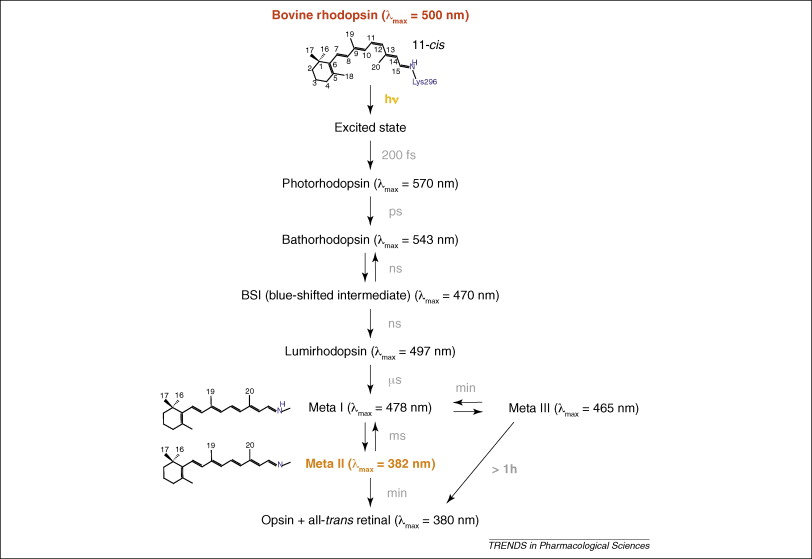 Upon absorption of light, one photon is enough, the retinal isomerizes to the trans form within 200 fs 36. Through a series of different intermediates it decays thermally (see figure)38. In the transition from Meta I to the Meta II intermediate the receptor undergoes a large conformational change to generate the G-protein-binding pocket on the intracellular side of the receptor. In 1996 Farrens et al. showed that there is an outward rotation of TM6 in the transition to Meta II37. This motion would create the cavity for the G protein to enter. The mechanism that connects the isomeration of retinal to the opening of the cavity remained illusive. This only began to be understood once the structures became available.
Rhodopsin was the first GPCR determined at high resolution. The reason crystals of this GPCR are easiest to obtain is because they can be isolated in large amounts from the retina's of cows.
The structures of inactive rhodopsin showed the 7TM bundle structure and revealed the location of residues that are conserved across the large A GPCR family.
There were several unexpected features of the structure. On the extracellular side, ECL2 was wedged between the TM helices and served as a cap on the retinal-binding site.
On the intracellular side, a short amphipathic helix was found to be oriented roughly perpendicular to the seven TM helices, termed (TM8), lying parallel to the membrane39.
The first clues to the structural changes occurring upon activation came from crystal structures of opsin that were determined with and without a bound undecapeptide mimic of the C-terminus of Gα. The defining feature of the opsin structure was the outward rotation of TM helix H6.
There are some interesting theories of how retinal isomeration is linked to the opening of the intracellular crevice. One of them states that the photon induced retinal isomeration triggers a chain reaction of molecular switches that involve most of the highly conserved residues. The rotation of the C20 methyl group toward ECL2 and motion of the β-ionone ring toward TM5 leads to a change in orientation of TM5. Tyr528 on the intracellular side of TM5 rotates toward Arg340 on TM3. Second, rotation of the retinal C20 methyl group and motion of the β-ionone ring enable Trp618 to rotate toward the extracellular surface. The motion of Trp618 triggers an internal switch involving Asn719, Met610 and Tyr733. Together, the reorientation of Tyr518 on H5, Met610 on H6 and Tyr733 on H7 stabilize the ionic lock in an ‘open’ conformation, allowing for G protein binding and activation38(Illustration).
Rhodopsin appears to have evolved mechanisms that stabilize the receptor not only in an inactive conformation in the dark but also in a fully active conformation on light absorption. The receptor is a robust on–off switch making use of light energy to bridge two very stable conformations.
Upon absorption of light, one photon is enough, the retinal isomerizes to the trans form within 200 fs 36. Through a series of different intermediates it decays thermally (see figure)38. In the transition from Meta I to the Meta II intermediate the receptor undergoes a large conformational change to generate the G-protein-binding pocket on the intracellular side of the receptor. In 1996 Farrens et al. showed that there is an outward rotation of TM6 in the transition to Meta II37. This motion would create the cavity for the G protein to enter. The mechanism that connects the isomeration of retinal to the opening of the cavity remained illusive. This only began to be understood once the structures became available.
Rhodopsin was the first GPCR determined at high resolution. The reason crystals of this GPCR are easiest to obtain is because they can be isolated in large amounts from the retina's of cows.
The structures of inactive rhodopsin showed the 7TM bundle structure and revealed the location of residues that are conserved across the large A GPCR family.
There were several unexpected features of the structure. On the extracellular side, ECL2 was wedged between the TM helices and served as a cap on the retinal-binding site.
On the intracellular side, a short amphipathic helix was found to be oriented roughly perpendicular to the seven TM helices, termed (TM8), lying parallel to the membrane39.
The first clues to the structural changes occurring upon activation came from crystal structures of opsin that were determined with and without a bound undecapeptide mimic of the C-terminus of Gα. The defining feature of the opsin structure was the outward rotation of TM helix H6.
There are some interesting theories of how retinal isomeration is linked to the opening of the intracellular crevice. One of them states that the photon induced retinal isomeration triggers a chain reaction of molecular switches that involve most of the highly conserved residues. The rotation of the C20 methyl group toward ECL2 and motion of the β-ionone ring toward TM5 leads to a change in orientation of TM5. Tyr528 on the intracellular side of TM5 rotates toward Arg340 on TM3. Second, rotation of the retinal C20 methyl group and motion of the β-ionone ring enable Trp618 to rotate toward the extracellular surface. The motion of Trp618 triggers an internal switch involving Asn719, Met610 and Tyr733. Together, the reorientation of Tyr518 on H5, Met610 on H6 and Tyr733 on H7 stabilize the ionic lock in an ‘open’ conformation, allowing for G protein binding and activation38(Illustration).
Rhodopsin appears to have evolved mechanisms that stabilize the receptor not only in an inactive conformation in the dark but also in a fully active conformation on light absorption. The receptor is a robust on–off switch making use of light energy to bridge two very stable conformations.
Ligand binding GPCRs
In contrast, the ligand-activated GPCRs have much smaller barriers to activation. Multiple receptor conformations can be populated, which leads to high basal activity, but also provides versatility in signalling and regulation.42,13 After the rhodopsin structure another seven years of extensive research and technology developments were needed to obtain the high-resolution structure of the human β2-adrenergic receptor (β2AR) – the first example of a GPCR with a diffusible ligand.47-49 That structure was followed by other class A (rhodopsin-like) GPCRs, including β1AR 50, A2A adenosine (A2AAR) 51, chemokine CXCR4 (CXCR4) 52, dopamine D3 (D3R) 53, histamine H1 (H1R) 54, sphingosine 1-phosphate (S1P1)55, M2 and M3 muscarinic acetylcholine(mAChR M2 and M3) 56,57, κ-opioid(κ-OR)58, µ-opioid(µ-OR)59, δ-opioid receptor(δ-OR)60 and the nociceptin/orphanin FQ (N/OFQ) peptide (NOP)61 receptors. The structures of these ligand binding GPCRs were either bound to an agonist, a partial agonist, an inverse agonist, a partial inverse agonist or an antagonist. One of them, a β2AR(PDBID: 3sn6.pdb), even has an entire G-protein bound.
Pharmacological definitions
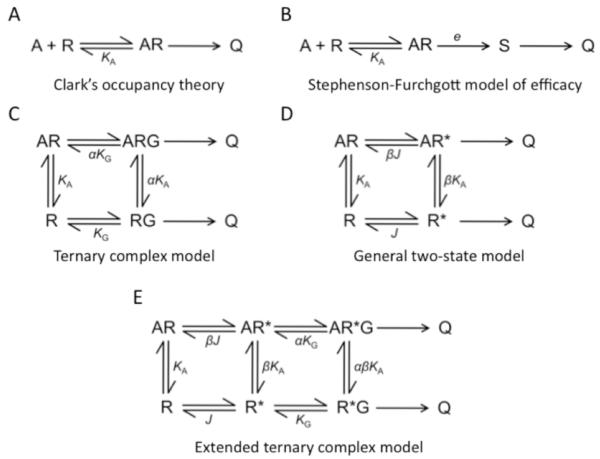
Ligands or drugs that interact with GPCRs are defined according to their activity when added to cells that contains the specific GPCR of interest. Different ligands can stabilize different structural conformations. Definitions of ligands according to their biological activity are listed below.

Agonist: a ligand that binds to and activates a receptor and elicits a physiological response. The endogenous agonist for the β1AR, noradrenaline, is a full agonist (Figure I, red line) that elicits the maximal response for the receptor in activating a G protein.
Basal or constitutive activity: a physiological response that occurs in the absence of an agonist or inverse agonist due to a proportion of the receptor being in the activated state.
Inverse agonist: a ligand that binds to a receptor and inhibits or eliminates, in the case of a full inverse agonist, the basal or constitutive activity of a receptor (Figure I, green line).
Neutral antagonist: a ligand that binds to the receptor, making it impossible for an agonist or an inverse agonist to bind, while maintaining the basal activity (Figure I, black line).
Partial agonist or weak partial agonist: a ligand that elicits only a partial response when compared to a full agonist (Figure I, blue and yellow lines).
Biased agonist: many ligands can differentially activate signaling pathways mediated via a single G protein-coupled receptor. Subsequent mechanisms that may play a role include diversity of G proteins, scaffolding and signaling partners, and receptor oligomers.68
These structures of different GPCRs in different stages of activation allowed researchers to look at the structural changes that occurred upon activation and compare the different GPCRs that were in the same state of activation. The general structure of the 7TM bundle appeared to be highly similar between the different GPCRs, which was predicted. The largest differences between the different GPCRs was of course the extracellullar part of the receptor, because they all have to bind to different ligands and be specific. When more and more of these structures were solved, a scala of hypotheses arose and the search for the main mechanism of activation had begun. In 2006 Schwartz et al.69 published a paper proposing a global toggle switch model for the activation mechanism to reconcile the accumulated biophysical data supporting an outward rigid-body movement of the intracellullar segments, as well as an recent data derived from activating metal ion sites and tethered ligands, which suggest an opposite, inward movement of the extracellullar segments of the TMs. According to this model a vertical see-saw movement of TM6, and to some degree TM7, around a pivot corresponding to the highly conserved prolines will occur during receptor actiavtion, which may involve the outer segment of TM5. Agonists can stabilize such a proposed active conformation, where the extracellullar segmenets of TM6 and TM7 are bent inward toward TM3, by acting as molecular glue deep in the main ligand-binding pocket between the helices whereas larger agonists, peptides and proteins can stabilize a similar active conformation by acting as Velcro at the intracellullar ends of the helices and the connecting loops. They proposed that the helices move in a see-saw-like manner. Recently Mason et al.70 used a series of computational methods of the X-ray structures elucidated so far and found that upon receptor activation, the volume of the ligand binding site decreases by ~40Å3 for the aminergic β1 and β2AR and the ~90Å3 for the purinergic A2A receptor; rhodopsin is the exception with an increase of ~100Å3. Indicating that agonists stabilize a receptor conformation in which the extracellullar sides of certain helices are closer together. This finding, combined with the finding of the see-saw-like motions of helices, results in model that could be compared to the mechanism that a clothespin uses.
Approach
We want to know what moves systematically upon activation of a GPCR, to elucidate the mechanism of GPCR activation.
At the moment (July 2012) there are 72 structures of GPCRs available from the Protein Data Bank (PDB). As mentioned a lot of these structures were modified to great extent.
 One way to compare all these GPCR structures is by superposing them based on their structure and look at the differences.(.sce)
One disadvantage of this approach is that it's impossible to draw conclusions from 72 superposed structures, like you can see in the figure.
One way to compare all these GPCR structures is by superposing them based on their structure and look at the differences.(.sce)
One disadvantage of this approach is that it's impossible to draw conclusions from 72 superposed structures, like you can see in the figure.
Another disadvantage is that superposition is relative like you can see in the picture below.
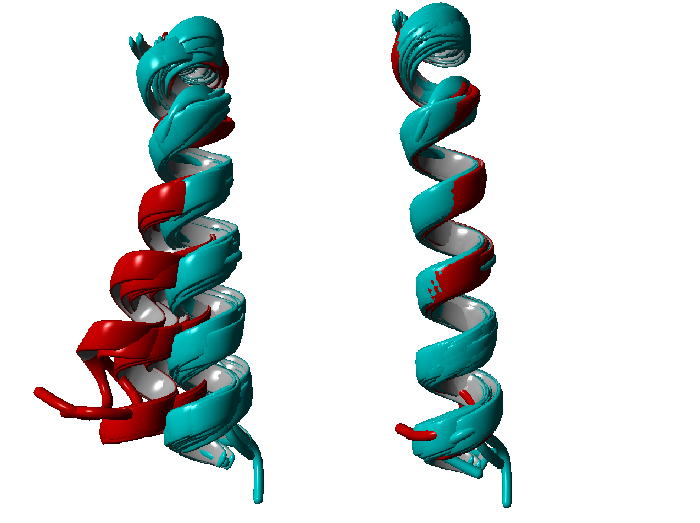 Both alignments are representations of TM6, red are the supposed active structures and in cyan are the supposed inactive structures. The left one is extracted from an alignment of the entire structures, which would lead to the conclusion that there is a bending of the transmembrane helix upon activation. The right one is a superposition of TM6 alone, which would lead to the conclusion that these is no significant difference between TM6 in the active and inactive state respectively. We decided to go into distance space, because distances are variables that are independent of 3D space.
We chose to use the Random Forest method to calculate which distances describe the difference between inactive and active structures the best.
Both alignments are representations of TM6, red are the supposed active structures and in cyan are the supposed inactive structures. The left one is extracted from an alignment of the entire structures, which would lead to the conclusion that there is a bending of the transmembrane helix upon activation. The right one is a superposition of TM6 alone, which would lead to the conclusion that these is no significant difference between TM6 in the active and inactive state respectively. We decided to go into distance space, because distances are variables that are independent of 3D space.
We chose to use the Random Forest method to calculate which distances describe the difference between inactive and active structures the best.
Random Forest
Random forest (RF) models 33 are non-parametric and non-linear models, attractive due to their interpretability.
They are based on averaging over a large collection of decision trees, each trained on a separate bootstrap sample of the input set.
The aggregate model has lower variance and is less susceptible to overfitting than a single decision tree. Gini Importance (GI) and Variable Importance (VI) are two measures of feature relevance that can be computed based on the RF model. 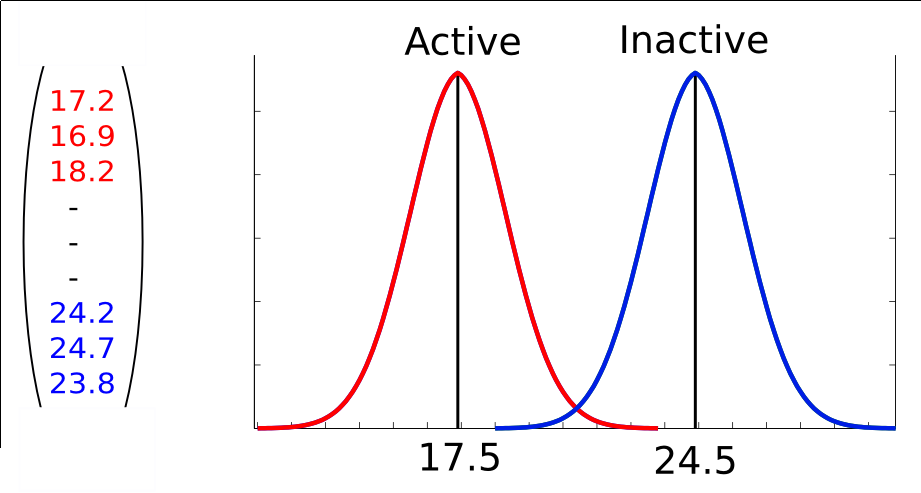 We use the R package randomForest 34 for training RF models. There are two parameters that influence the performance of RF: the number ntree of trees in the collection and the number mtry of variables considered for each tree split. In our experiments, we use the recommended value of mtry (square root of number of features).
We use the R package randomForest 34 for training RF models. There are two parameters that influence the performance of RF: the number ntree of trees in the collection and the number mtry of variables considered for each tree split. In our experiments, we use the recommended value of mtry (square root of number of features).
In 2001 Breiman et al. proposed random forests, which add an additional layer of randomness to bagging. In addition to constructing each tree using a different bootstrap sample of the data, rondom forests change how the classifiction or regression trees are constructed.34 Random forests are becoming increasingly popular in many scientific fiels because they can cope with "small n large p" problems, complex interaction and even highly correlated predictor variables.67
Each tree is built as follows33: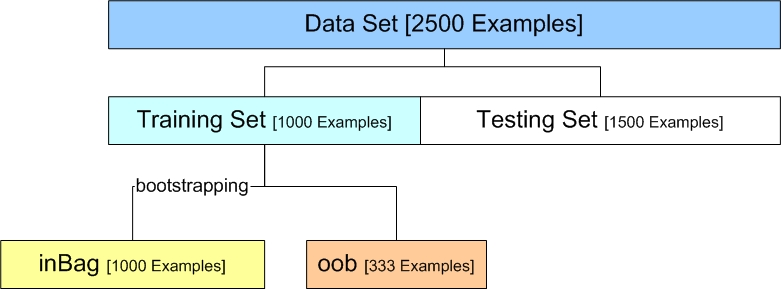
1) If the number of cases in the training set is N, sample N cases at random - but with replacement, from the original data. This sample will be training set for growing the tree.
2) If there are M input variables, a number m<<M is specified such that at each node, m variables are selected at random out of the M and the best split of these m is used to split the node. The value of m is held constant during the forest growing.
3) Each tree is grown to the largest extent possible. There is no pruning.
Some important features, that the RF method has, are that it runs efficiently on large data bases, it can handle thousands of input variables without variable deletion and it gives estimates of what variables are important in the classification. The latter will be extensively discussed below.
Variable Importance
This is a difficult concept to define in general, because the importance of a variable may be due to its (possibly complex interaction with other variables. The random forest algorithm calculates the importance of a variable using the out-of-bag individuals according to the following logic: If randomly permuting values of a variable does not affect the predictive ability of trees on out-of-bag samples, that variable is deemed as unimportant. If the variable drastically impairs the ability of trees to correctly predict the class of out-of-bag samples after permutation, that variable is given a high importance score. This measure of variable importance is called "permutation importance". In addition to this measure the algorithm calculates another measure of variable importance called "Gini importance", which is calculated as follows: Every time a split of a node is made on variable m the gini impurity criterion for the two descendent nodes is less that the parent node. Adding up the gini decreases for each individual variable over all trees in the forest gives a fast variable importance that is often very consistent with the permutation importance measure. 33,34,66
