Articles
EU name: WIF009
(Date: Aug 14 2018 WIF009 )
WHAT_CHECK is part of the WHAT IF project.
There are a large number of articles associated with the WHAT IF project.
At the release date of WHAT IF 6.0 (1-1-2006)
there were more than 1300 references
to the WHAT IF article, and more than 500 to the WHAT_CHECK article, in
the literature. And we hope that you don't
forget to refer to WHAT IF and WHAT_CHECK in your work.
WHAT IF
If you use WHAT IF, please refer to:
WHAT IF: A molecular modeling and drug design program.
G.Vriend, J. Mol. Graph. (1990) 8, 52-56.
WHAT_CHECK
If you use WHAT_CHECK, please refer to:
Errors in protein structures.
R.W.W. Hooft, G. Vriend, C. Sander, E.E. Abola, Nature (1996) 381, 272-272.
WHAT IF servers
If you used one of the WWW-based servers, please refer to:
Homology modeling, model and software evaluation: three related
resources.
R.Rodriguez, G.Chinea, N.Lopez, T,Pons, G.Vriend CABIOS (1998) 14, 523-528.
WHAT IF technologies
The WHAT IF project has been used to implement a series of ideas.
The article section of the WHAT IF
pages describe those ideas.
WHAT_CHECK technologies
The following list of articles describes some of the WHAT_CHECK checks:
The QUALTY menu
The quality menu was
WHAT IF's first step on the long path
of protein structure validation.
This article describes the packing quality control module of
WHAT IF. This method is also called DACA for Directional Atomic
Contact Analysis. The idea is that the distribution of atom
types is determined around amino fragments. We assume that
the average distribution observed in the PDB is representative
for what can happen in nature. Unlike many other methods, we
do not spherically average the distribution around the fragment,
but we keep the x,y,z directionality of the contacts. The
quality of any structure is now easily determined by a
convolution of the average distributions and the observed
contacts in the protein to be checked.
Quality control of protein models: Directional atomic
contact analysis.
G. Vriend, C. Sander. J.Appl.Cryst. (1993) 26, 47-60.
(PDF).
The MUTATE etc menu
WHAT IF has been used extensively to predict point mutations
in proteins. The idea (free after Jones and Thirup) is to
extract from the database fragments that fit well on the fragment
that has the residue to be mutated in the middle. If the fragments
are selected to only have the desired (novel) residue in the middle
one obtains a so-called position specific specific rotamers.
These next three siles, extracted from an old homology
modelling seminar, illustrate that:
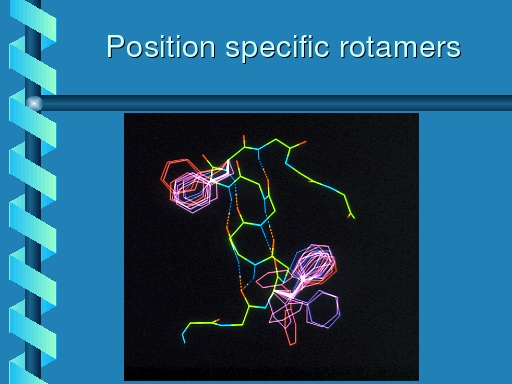
|
Rotamer distribution for phenylalanine at two positions in
a protein. The one position clearly has a preference for
one of the three primary rotamers; at the other position there
also seems to be one favourite, but the preference is slightly
less strong.
|
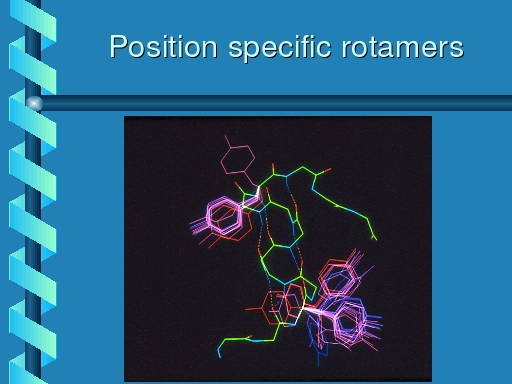
|
Rotamer distributions for tyrosine at the same two positions.
The top left rotamer shows the same distribution as observed for
phenylalanine, but the bottom right one is much more bimodal.
|
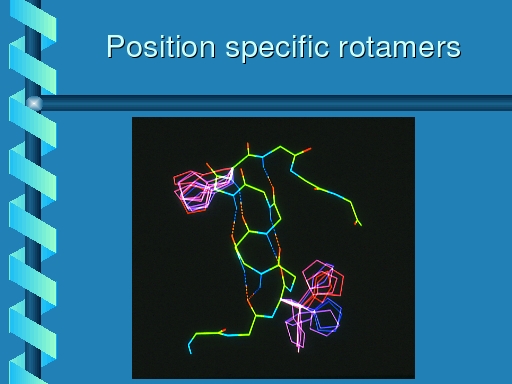
|
Rotamer distributions for histidine at the same two positions.
The top left rotamer shows the same distribution as observed for
phenylalanine, and tyrosine, but the botton right one now is equally
populated in each of its three primary rotamers.
|
This work was published in:
The use of position specific rotamers in model building by
homology.
G. Chinea, G. Padron, R.W.W.Hooft, C.Sander, G.Vriend,
PROTEINS (1995) 23, 415-421.
The CHECK menu
All protein structures solved by NMR or X-ray (that have been
deposited in the PDB) contain errors ranging from small problems
like bond lengths that are a bit too long till catastrophical
things like backwards threaded sequences.
Rob Hooft wrote most options of the
CHECK menu in WHAT IF. The first
thing published was the software to reconstruct the full cell from
the cell and scale information provided in the PDB file. It is
amazing how many different errors the crystallographers can make
in so few information:
Reconstruction of symmetry related molecules from protein data
bank (PDB) files.
R. Hooft, C. Sander, G. Vriend, J. Appl. Cryst. (1994) 27,
1006-1009.
An important aspect of the CHECK facility is the option to get
all hydrogenbonds right. More than 10% of all asparagines,
glutamines, and histidines in the PDB are flipped 180 degrees
about their chi-2, chi-3, and chi-2 side chain torsion angles,
respectively. The hydrogenbond optimization software finds these
problems:
Positioning hydrogen atoms by optimizing hydrogen-bond
networks in protein structures.
R.W.W.Hooft, C.Sander, G.Vriend, PROTEINS (1996) 26, 363-376.
Side chain planarity has been close to random in the early
days of X-ray refinement and even today there are still authors
of well-known refinement packages (e.g. D.T. and A.B.) who
think they know better than the results that can be
extracted from a study of the CSD. Anyway, structures refined
with those softwares are invariably flagged by:
Verification of protein structures: Side-chain planarity.
R.W.W. Hooft, C.Sander and G.Vriend,
J. Appl. Cryst. (1996) 29, 714-716.
This paper describes the verification of side-chain planarity
by WHAT IF and WHAT_CHECK. It also describes the construction of
a representative list of PDB files as used in the WHAT IF database.
This latter facility is available as the
../select/
PDBSELECT database.
The Ramachandran plot is probably the best determinant of protein
quality. This was first realized by Thornthon and Laskowsky. Others
(e.g. T.A.J.) later tried to improve their PROCHECK software by
tightening the borders a bit. Rob's approach to do things with
proper statistics and with residue specificity, however, was a
significant improvement:
Objectively judging the quality of a protein structure
from a Ramachandran plot.
R.W.W. Hooft, C.Sander and G.Vriend, CABIOS (1997), 13, 425-430.
The X-ray cell dimension validation of WHAT IF is decribed in:
Some WHAT_CHECK checks explained.
R.W.W.Hooft, G.Vriend, PDB Newsletter. (1998) April volume.
The quality of the quality checks was explained in:
Who checks the checkers? Four validation tools applied to
eight atomic resolution structures.
K.Wilson, C.Sander, R.W.W.Hooft, G.Vriend, et al.
J.Mol.Biol. (1998) 276,417-436.
Validating protein structures (and models) is one of our
hobbies. This article (which has 20 authors, the whole validation
consortium is on it...) describes the results of validating some
structures solved at atomic resolution (around 1.0 A) that thus
were supposed to be guaranteed correct. This study revealed some
problems in the ultra highly refined structures and some problems
in the refinement programs. The general conclusion should be that
the validation programs are actually working very well, and that
remarks by some crystallographers that "these validation programs
give very many false positives" really are not supported by
experimental verification.
Rob Hooft mainly wrote X-ray specific validation options, and he wrote
the WHAT IF infra-structure for validation in general. Other
people have worked on NMR structure validation. Jurgen Doreleijers
has for many years worked on getting NMR ensembles simply
administratively correct. A mamoth task...
Validation of NMR structures of proteins and nucleic acids:
hydrogen geometry and nomenclature.
J.F.Dorelijers, G.Vriend, M.L.Raves, R.Kaptein
Proteins (1999) 37, 404-416.