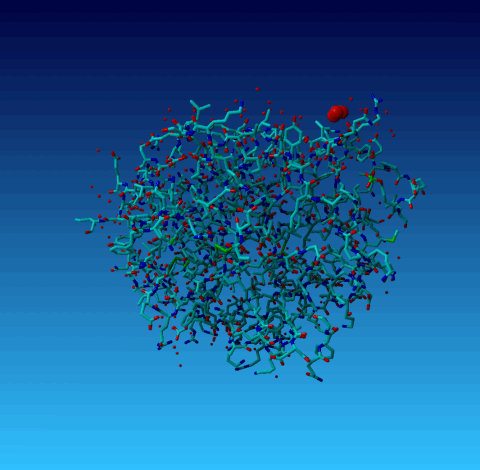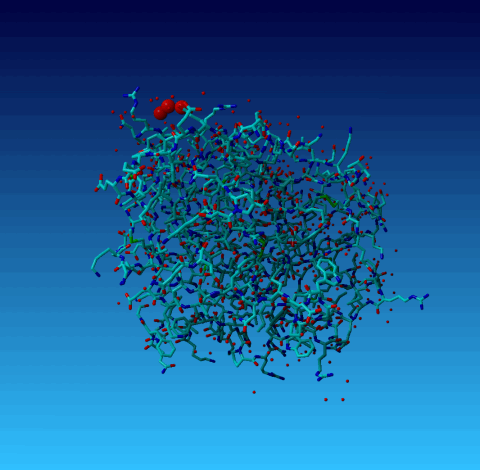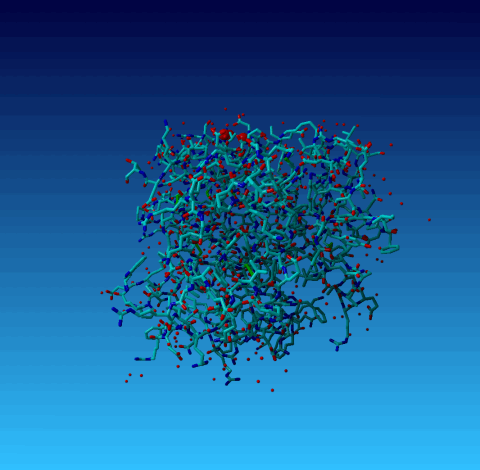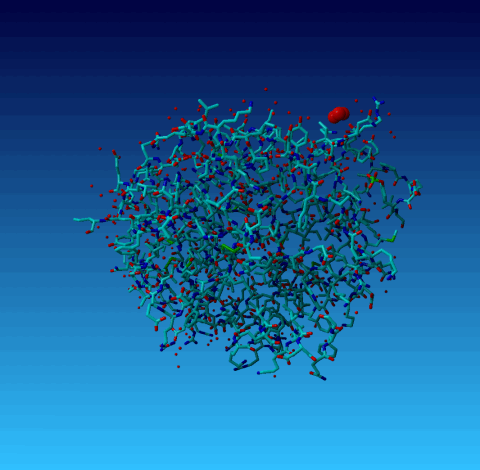
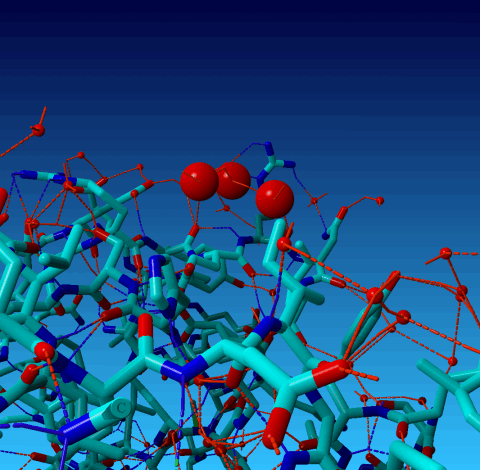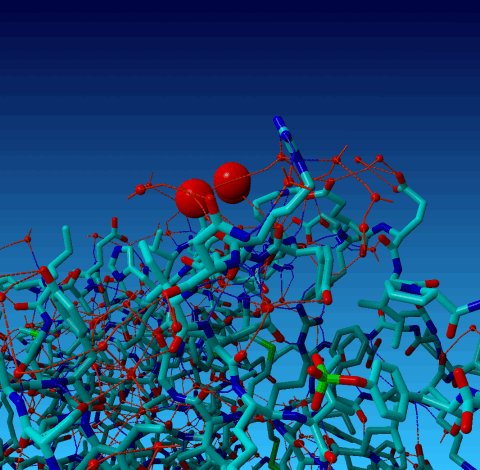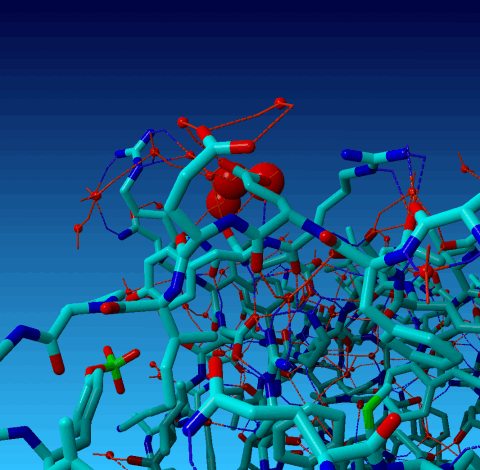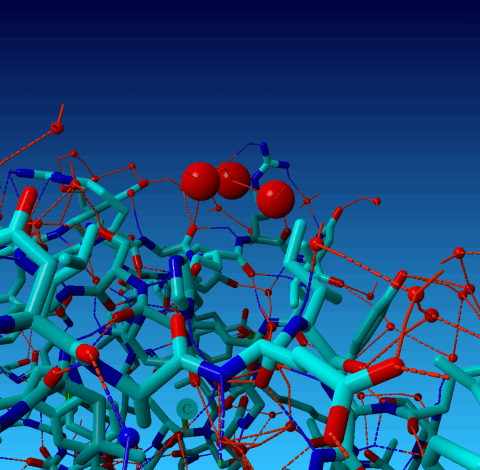
Figure 18. Normal YASARA stick view of 1AHT. Waters are shown as red spheres. The three waters 901, 902, and 903 are big balls.
Section three of the validation report deals with Occupancies and B-factors. These are typical X-ray topics as in NMR structure solution the occupancies are dealt with through the ensemble distribution.
In this section we look for missing atoms. There is nothing wrong with missing atoms in X-ray structures, but in NMR structures it is an error. However, if very many atoms are missing, the end-user of the PDB file might decide to
If alternate atoms are observed, than the occupancies of the alternates should add up to 1.0 in most cases, and to a lower number in case there are yet even more rotamers that are occupied, but that do not give a strong enough signal to lift them out of the noise. In such cases we issue a warning:
Alternate atom occupancy Warning: Occupancies atoms do not add up to 1.0. |
The PDB uses coordinates that are written with the format F8.3. That means that there are three digits behind the decimal point. So, (-3.124, 17.186, 0.224) are valid coordinates for an atom. Obviously, the third decimal is meaningless, and in a file like 1AC8 all coordinates have a zero as the last decimal.
JRNL AUTH M.M.FITZGERALD,R.A.MUSAH,D.E.MCREE,D.B.GOODIN JRNL TITL VARIATION IN STRENGTH OF AN UNCONVENTIONAL C-H TO JRNL TITL 2 O HYDROGEN BOND IN AN ENGINEERED PROTEIN CAVITY JRNL REF J.AM.CHEM.SOC. V. 119 626 1997 |
However, in this same file we also find a few residues that have the last two decimals zero for some atoms:
ATOM 1 N LEU A 4 13.030 92.360 76.550 1.00 38.40 N ATOM 2 CA LEU A 4 12.200 92.700 75.400 1.00 35.28 C ATOM 3 C LEU A 4 11.600 91.520 74.650 1.00 33.10 C ATOM 4 O LEU A 4 12.340 90.720 74.080 1.00 33.13 O ATOM 5 CB LEU A 4 12.970 93.520 74.370 1.00 37.04 C ATOM 6 CG LEU A 4 12.820 95.010 74.400 1.00 37.38 C ATOM 7 CD1 LEU A 4 13.590 95.650 73.270 1.00 37.59 C ATOM 8 CD2 LEU A 4 11.340 95.350 74.230 1.00 41.18 C ATOM 9 H LEU A 4 12.630 92.150 77.420 1.00 0.00 H |
I guess that this is to be expected as 1/1000 of all atoms should have the two last decimals of all three coordinates at zero when all third digits are zero.
JRNL AUTH Z.CHEN,Y.LI,A.M.MULICHAK,S.D.LEWIS,J.A.SHAFER JRNL TITL CRYSTAL STRUCTURE OF HUMAN ALPHA-THROMBIN JRNL TITL 2 COMPLEXED WITH HIRUGEN AND P-AMIDINOPHENYLPYRUVATE JRNL TITL 3 AT 1.6 A RESOLUTION. JRNL REF ARCH.BIOCHEM.BIOPHYS. V. 322 198 1995 |
In 1AHT we find three water atoms with 'funny' coordinates:
HETATM 2597 O HOH H 901 0.000 4.700 0.000 1.00 34.53 O HETATM 2598 O HOH H 902 0.000 0.500 0.000 1.00 33.40 O HETATM 2599 O HOH H 903 0.100 2.200 -1.100 1.00 29.87 O |
WHAT_CHECK warns for two of them that they are located at a special position, and it warns that they have rounded coordinates. These two things, obviously, are related. So lets look at it:
|
|
Figure 18. Normal YASARA stick view of 1AHT. Waters are shown as red spheres. The three waters 901, 902, and 903 are big balls. |

|
Figure 19. Same picture as the previous one, but now the outer cell axes are shown so you can see that two of the waters are essentially on an axis, and one is very near to it. |
|
Figure 20. Same picture again, but now the optimized H-bond network is shown. You see that these three waters each make only one, not very strong, H-bond with the protein. So, in summary, i think that these waters are placed in Fourier ripples or something like that, and not in genuine density. This cannot be checked because the 1AHT reflections have not been deposited. |
JRNL AUTH S.BARANIDHARAN,W.J.RAY JR.,Y.LIU JRNL TITL BINDING DRIVEN STRUCTURAL CHANGES IN CRYSTALINE JRNL TITL 2 PHOSPHOGLUCOMUTASE ASSOCIATED WITH CHEMICAL JRNL TITL 3 REACTION JRNL REF TO BE PUBLISHED |
In 1c47 we find funny waters with rounded coordinates. In the supplemental material I list all waters. I see no correlation between B-factors and coordinate rounding, albeit that there is a funny block of waters somewhere in the middle of the list that all got the same artificial B-factor of 25.0. These I coloured red.
Supplemental material
JRNL AUTH M.SHOHAM,A.YONATH,J.L.SUSSMAN,J.MOULT,W.TRAUB, JRNL AUTH 2 A.J.KALB JRNL TITL CRYSTAL STRUCTURE OF DEMETALLIZED CONCANAVALIN A: JRNL TITL 2 THE METAL-BINDING REGION. JRNL REF J.MOL.BIOL. V. 131 137 1979 |
In 1CN1 all coordinates are rounded. That is caused most likely by the fact that in the late 70's many structures were still solved using metal amino acids that were screwed together in a Richards box.

|
Figure 21. Picture of a Richards box. On the top you see the plastic sheets with drawn density contours and the half mirrors, on the bottom the molecule as metal frame. Ps, I do not know which molecule is in this picture. It is not 1CN1. The picture was obtained from Fred Richards obituary blogspot. |
Going through the list of all files with rounded coordinates, I see many very old PDB files, or files solved with 'other' techniques such as electron diffraction.
JRNL AUTH D.J.NEIDHART,P.L.HOWELL,G.A.PETSKO,V.M.POWERS, JRNL AUTH 2 R.S.LI,G.L.KENYON,J.A.GERLT JRNL TITL MECHANISM OF THE REACTION CATALYZED BY MANDELATE JRNL TITL 2 RACEMASE. 2. CRYSTAL STRUCTURE OF MANDELATE JRNL TITL 3 RACEMASE AT 2.5-A RESOLUTION: IDENTIFICATION OF JRNL TITL 4 THE ACTIVE SITE AND POSSIBLE CATALYTIC RESIDUES. JRNL REF BIOCHEMISTRY V. 30 9264 1991 |
In 2MNR we find again rounded coordinates. This time for four water molecules that are all located at special positions and have occupancy 1.0. When this file gets re-refined (in the PDB_REDO suite), we see that those four waters have gotten an occupancy of 0.5. This is undoubtedly one of the reasons that the PDB_REDO file is so much better than the original one:
| From PDB header | Calculated from data | After re-refinement | |
|---|---|---|---|
| R | 0.1620 | 0.1512 | 0.1400 |
| R-free | 0.2000 | 0.1530 | 0.1702 |
| σR-free | 0.0026 | 0.0029 | |
| R-free Z-score | 10.35 | -1.24 |
If you ask software to put a residue in a molecule and you just ask for standard coordinates, you are likely to get side chain torsion angles that are exact multiples of 60 degrees. Often such angles indicate that a side chain was modelled and that the coordinates are not based on density.
JRNL AUTH A.J.OAKLEY,M.LO BELLO,G.RICCI,G.FEDERICI,M.W.PARKER JRNL TITL EVIDENCE FOR AN INDUCED-FIT MECHANISM OPERATING IN JRNL TITL 2 PI CLASS GLUTATHIONE TRANSFERASES. JRNL REF BIOCHEMISTRY V. 37 9912 1998 |
In 16GS both copies of Asn 57 (once in A-chain, once in the B-chain) have a χ-1 angle of exactly 180.0 degrees. Obviously, this can happen by accident. After all, if the χ-1 angle was exactly 176.124 degrees, I would not have complained. But the funny thing is that the RMS coordinate difference between the two chains is about 0.1 Ångström, the B-factors of these two asparagines are normal (even a bit lower than the residues around it), and if we re-refine the structure we get similar, but not identical and not exactly 180.0 degree, torsion angles.
Amino acid # chain φ ψ Ω χ-1 χ-2 Original PDB file 56 ASP ( 57 ) A -129.3 92.9 -177.0 180.0 -4.2 264 ASP ( 57 ) B -130.8 92.4 -176.9 180.0 -4.7 Re-refined PDB file from PDB_REDO 56 ASP ( 57 ) A -124.8 97.5 -164.9 177.8 -1.9 264 ASP ( 57 ) B -129.9 97.4 -167.7 -177.9 -5.7 |
Summary, I have no idea what happened. Caca passa?
While browsing through some of the 2500 or so files that have this problem one way or another, I find many NMR files. That makes sense because in NMR one can just as well have missing data as in X-ray, but in the NMR community it is common practice to deposit all atoms regardless whether they have experimental backing or not. I also find a series of ligands. Like the arginine in an arginine repressor (1B4B), or asparagine in asparagine synthetase (11AS). In these cases we have to assume that either the refinement software couldn't cope with free amino acids, or something like that.
In any case, when the side chain torsion angles are a very precise multiple of 60 degrees, there is always something funny going on.