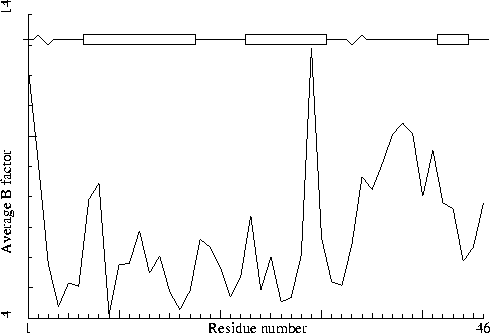
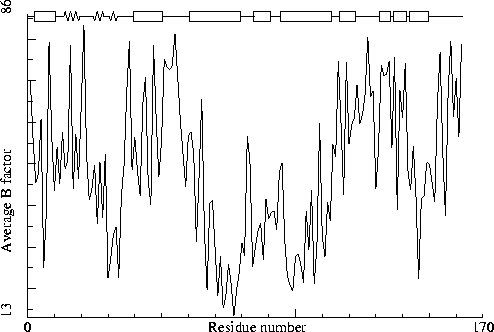
Crambin. PDB-id=1CRN.
The B-factor plot was what gave away the fraud by Murthy, so these plots seem important.
To illustrate this I will first list a series of B-factor plots for real structures (not entirely randomly chosen, these structures were selected based on A) I have worked with these structures in one project or the other; and B) I personally know the main author of the PDB entry, and trust him/her).
|
|
Crambin. PDB-id=1CRN. |

|
|
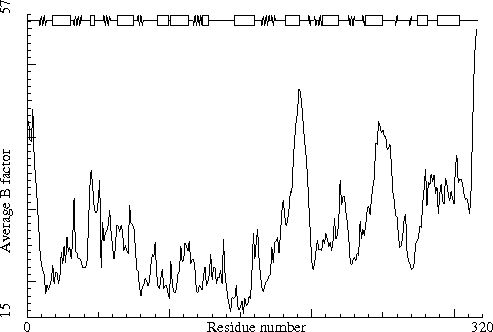
Octamer motif. PDB-id=1OCT. B-chain
|
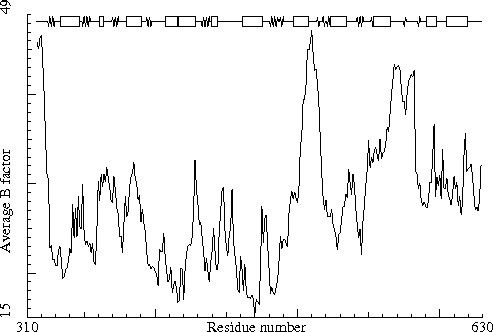
Sugar binding protein. PDB-id=2PZM. A-chain. |
|
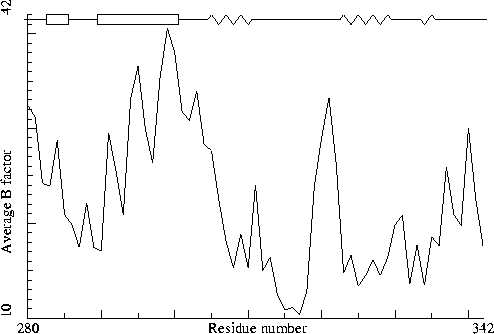
Sugar binding protein. PDB-id=2PZM. B-chain. |

|
Thermitase-eglin complex. PDB-id=3TEC. Enzyme. |
|
Thermitase-eglin complex. PDB-id=3TEC. Inhibitor. |
|
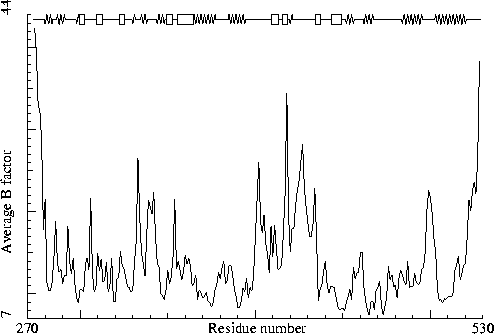
Rhino-14 capsid protein VP2. PDB-id=4RHV. (I solved this one myself!) |
|
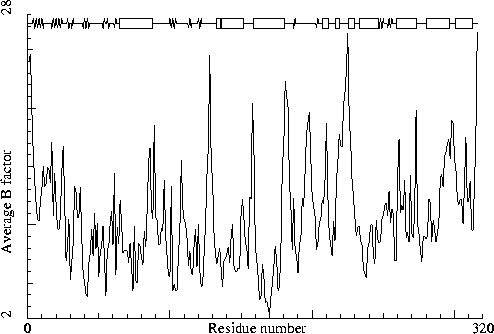
Thermolysin. PDB-id=5TLN. |

|
PDB-id=2qid B-chain |
The ones below I plot with a different program that plots B-factor plots for the whole PDB file rather then one molecule at a time, and that has the vertical axis start at zero rather than a bit below the lowest value. This shows the funny behaviour of Murthy's B-factors even better.

|
PDB-id=1bef |
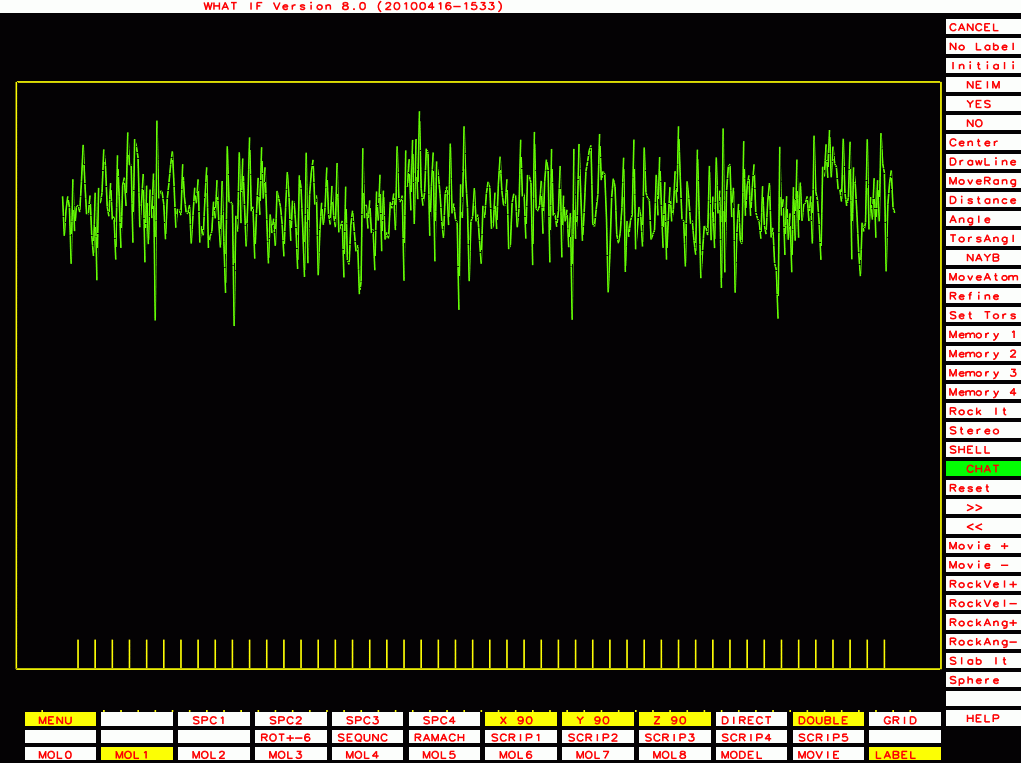
|
PDB-id=1g40 |
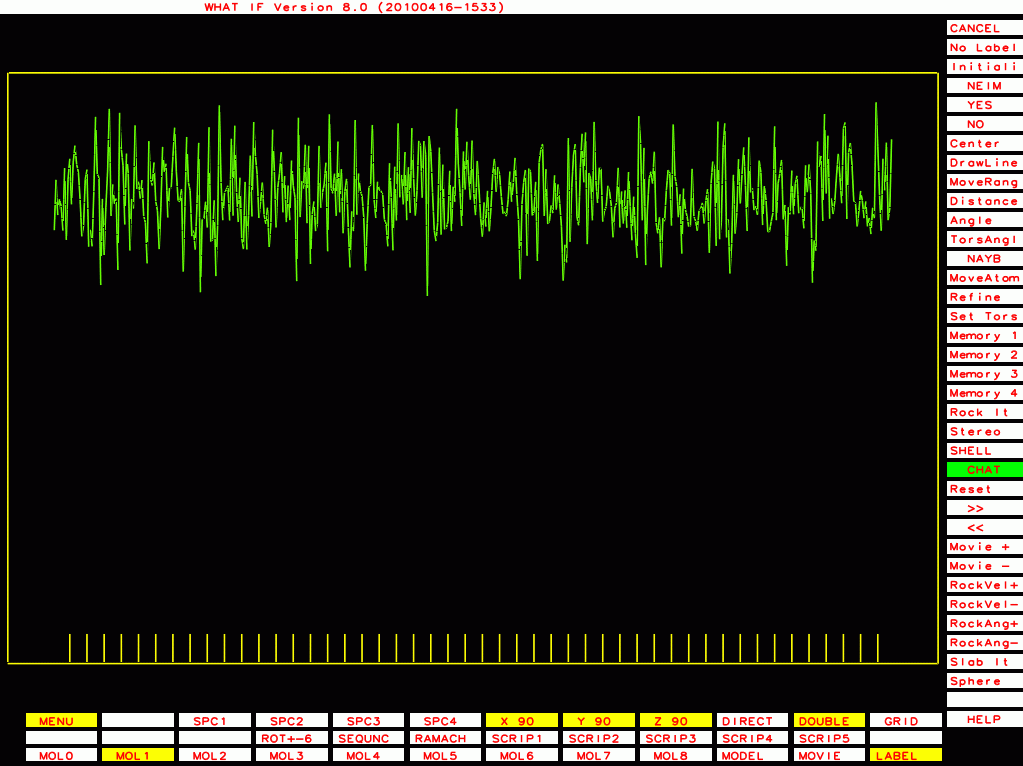
|
PDB-id=1rid |
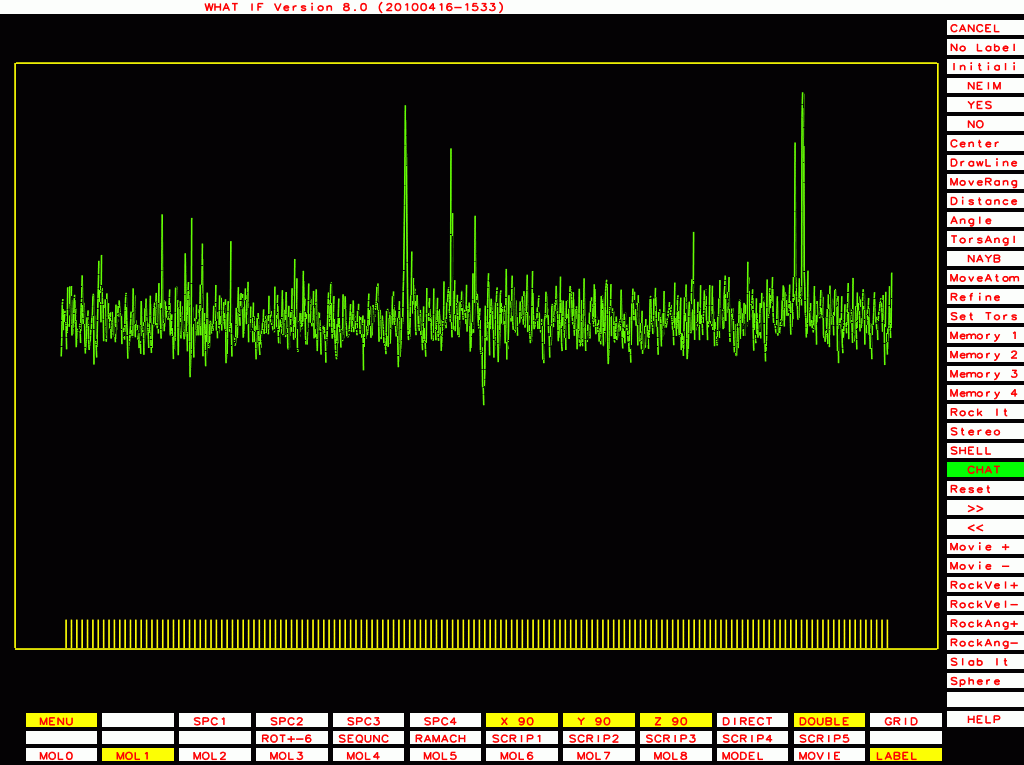
|
PDB-id=2hr0 |
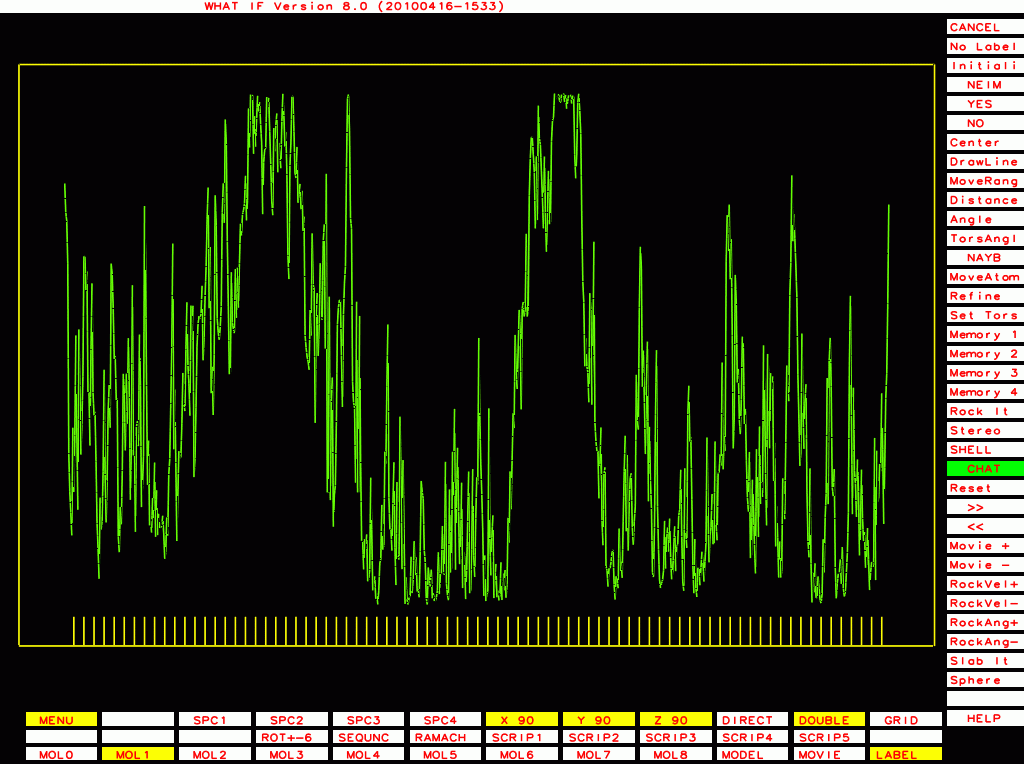
|
PDB-id=1cmw |
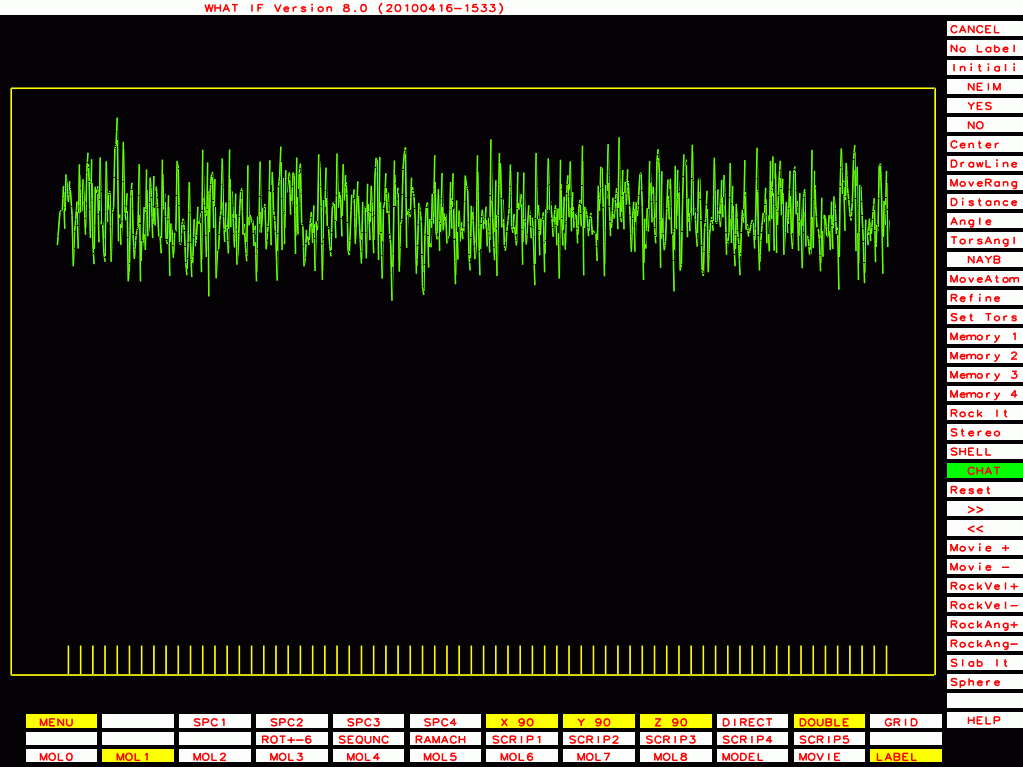
|
PDB-id=1g44 |
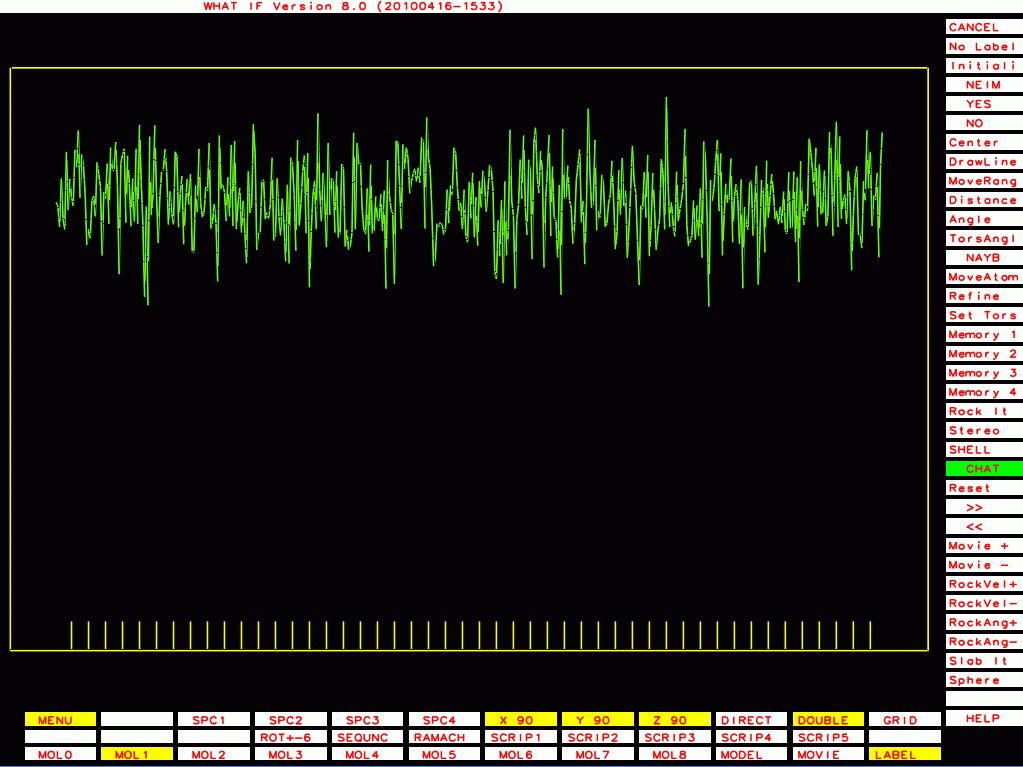
|
PDB-id=1y8e |
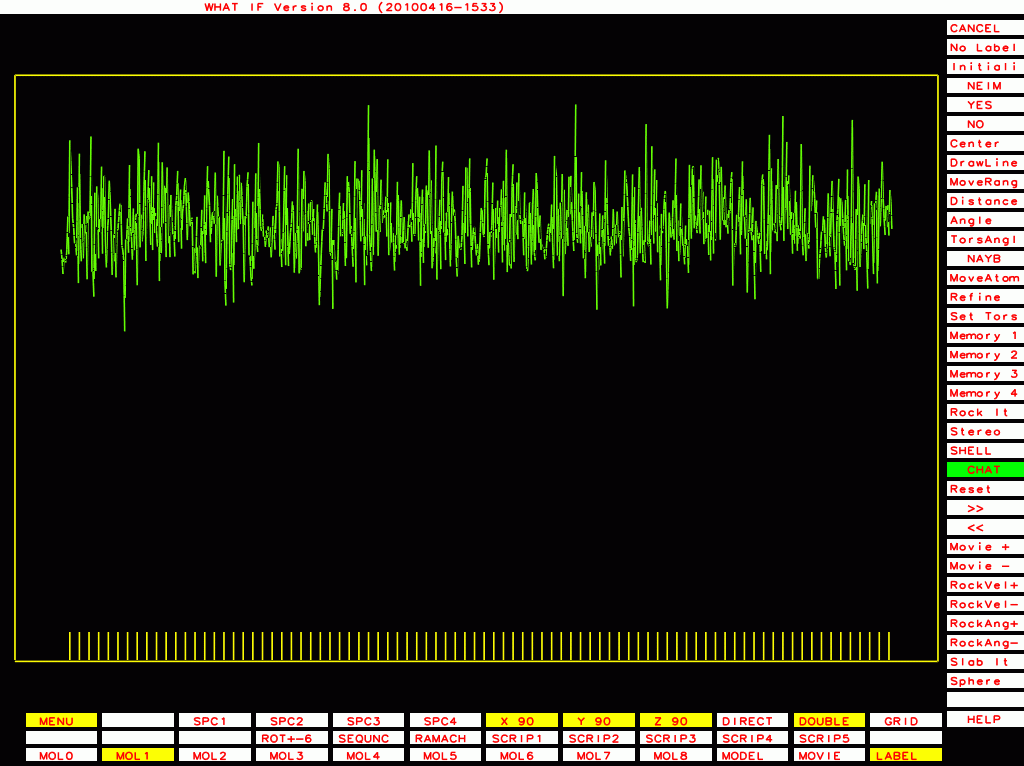
|
PDB-id=2ou1 |

|
PDB-id=1df9 |
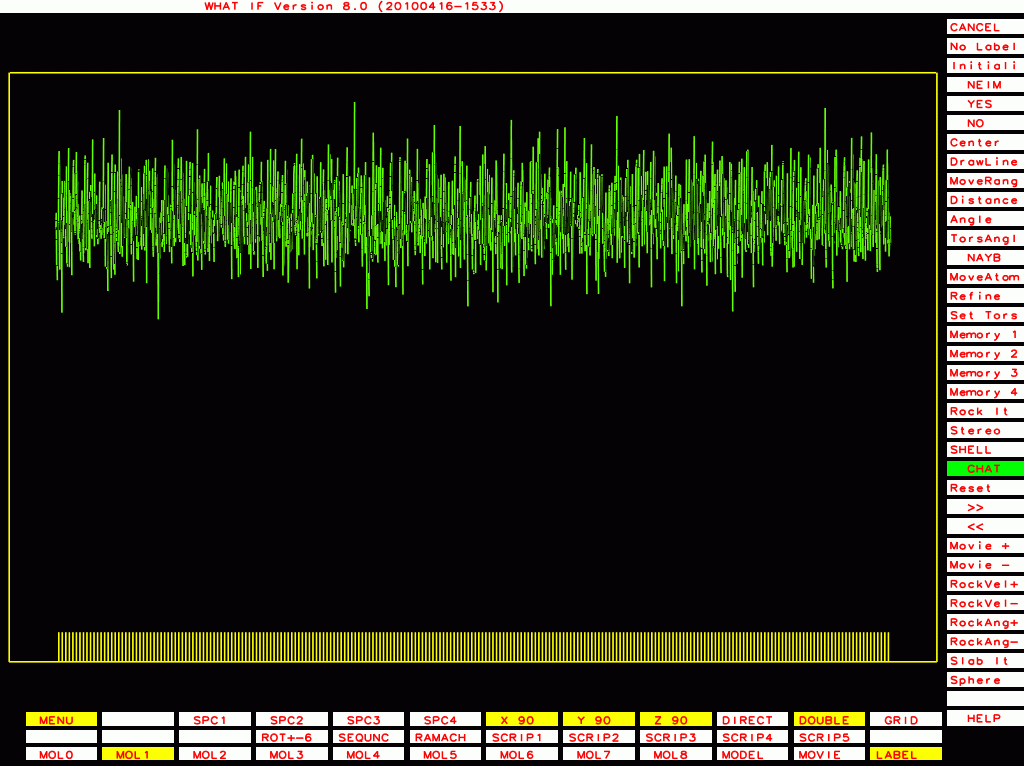
|
PDB-id=1l6l |
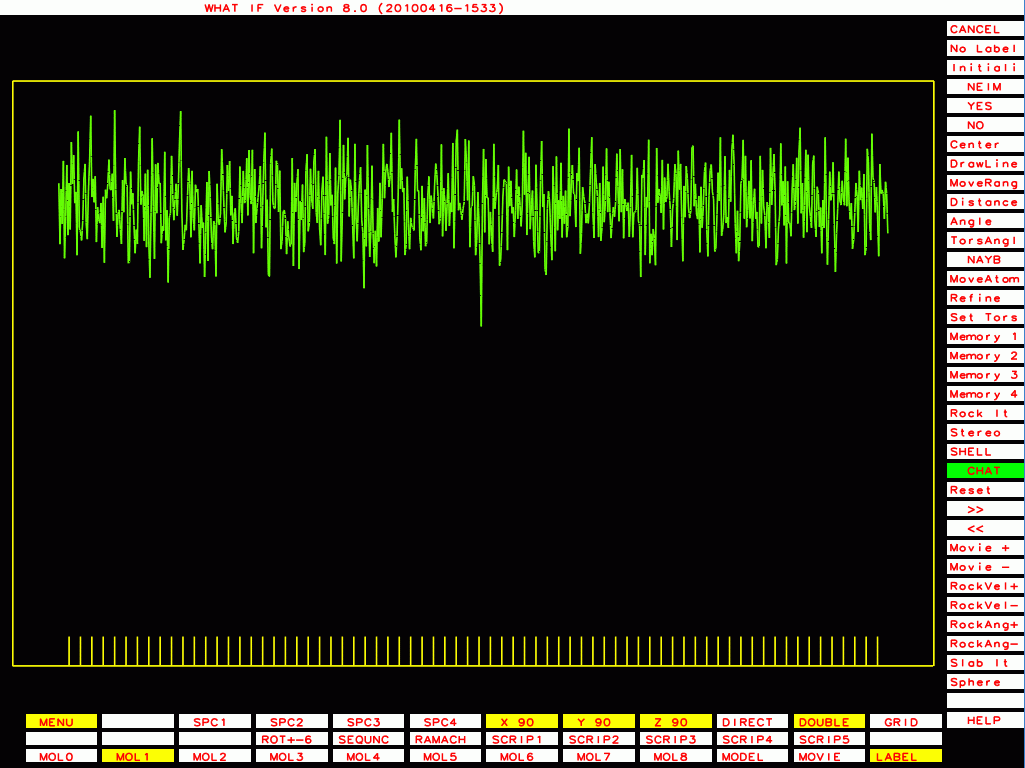
|
PDB-id=2a01 |
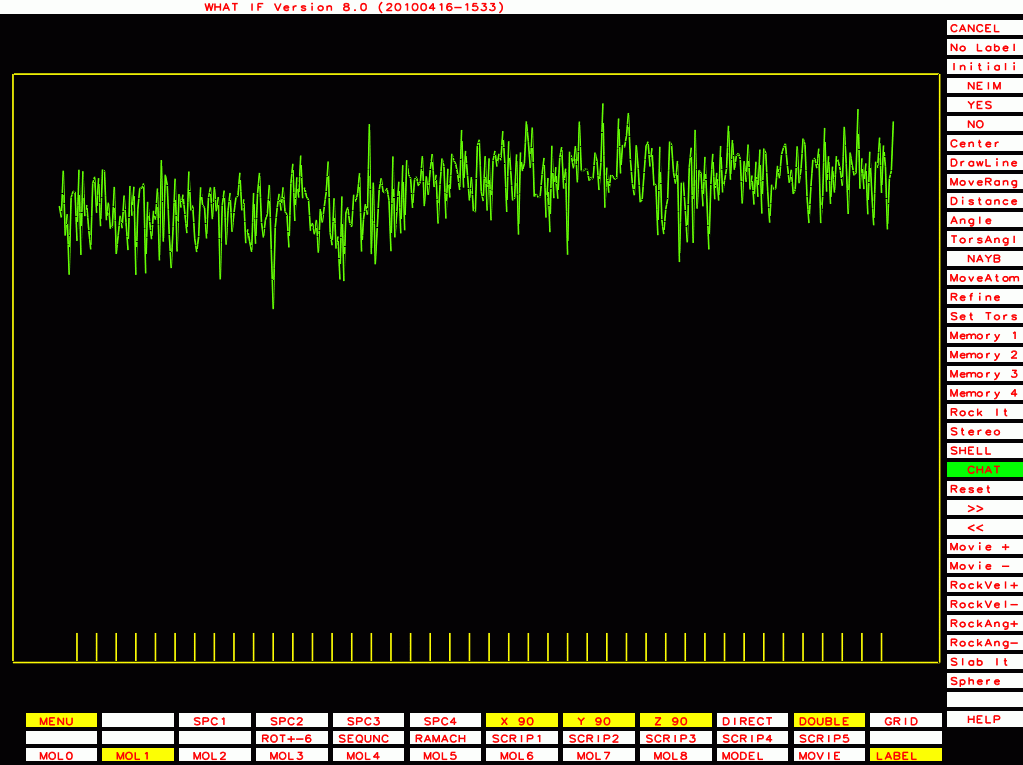
|
PDB-id=2qid |
WHAT_CHECK calculates an M-factor that expresses this weird B-factor distribution. The algorithm is explained in the:
Supplemental materialM-factors tend to be 0.2-0.4 or so for 'normal' PDB files. M-factors tend to be close to zero in Murthy files. We report the M-factor when it gets below 0.1. In July 2010 we looked at all M-factors in the PDB and found about 30 files with such an M-factor. In about half of the cases nothing seemed wrong, while in several cases there was an obvious reason for this behaviour.
Be aware that TLS can do things to B-factors that I don't understand. I do not know if this can have an influence on the M-factor.
It seems obvious that two atoms that are covalently bound to each other cannot have widely different B-factors. Still this occasionally happens. We have found situations (especially in structures refined with one particular refinement program...) where the Cγ in phenylalanine has a higher B-factor than the Cβ and the two Cδs it is covalently bound to.
If the B-factor distribution seems too improbable in terms of differences between covalently bound atoms you get the message:
B factor Z-score Error: The B-factors of bonded atoms show signs of over-refinement |
If you are a crystallographer solving a crystal structure you can consider limiting the freedom of your B-factors in refinement by tightening some parameter, or perhaps you should give in each side chain all atoms the same B-factor. Perhaps you should even consider not refining individual B-factors at all. It also seems wise to talk with an experienced crystallographer about using TLS instead of individual B-factors.
In 'normal' X-ray structures you do expect buried atoms to have a lower B-factor, on average, than atoms that are located at the surface. But individual exceptions are commonly observed. First, not all surface atoms are at the surface in the crystal (crystal packing reduces the freedom of the surface residues involved). Second, most proteins have a few buried residues with higher B-factor than 'the rest'. If we do not observe this, we issue a warning:
B factor Z-score Error: The B-factors of bonded atoms show signs of over-refinement |
Things can also go the other way around. In the report for 151L we find as B-factor plot:
|
Figure 22. The B-factor plot for 151L. |
and the warning text:
Warning: Average B-factor problem The average B-factor for all buried protein atoms normally lies between 10--20. Values around 3--5 are expected for X-ray studies performed at liquid nitrogen temperature. Because of the extreme value for the average B-factor, no further analysis of the B-factors is performed. Average B-factor for buried atoms : 38.587 |
The structure 151L is probably correct in most of its characteristics. And it is definitely a very good structure, especially given the fact that it was deposited in 1994. But the funny B-factor plot tells us that something weird has happened. Re-refining this structure in 2009 did not improve things very much, so perhaps the crystals weren't very good? We don't know. So, that is why WHAT_CHECK issues a warning.
It seems obvious that the B-factor of the Cγ of a phenylalanine can not be higher than the B-factor of its direct neighbours. And it seems unlikely that the Cδs of a leucine will have B-factors around 70 when the B-factor of the Cβ is around 14 and the B-factor of the Cγ is 21 or so. Still we often observe such problems; especially in structures refined with TNT.
In 141L we observe such a phenylalanine anyway:
JRNL AUTH E.P.BALDWIN,O.HAJISEYEDJAVADI,W.A.BAASE, JRNL AUTH 2 B.W.MATTHEWS JRNL TITL THE ROLE OF BACKBONE FLEXIBILITY IN THE JRNL TITL 2 ACCOMMODATION OF VARIANTS THAT REPACK THE CORE OF JRNL TITL 3 T4 LYSOZYME. JRNL REF SCIENCE V. 262 1715 1993 |
ATOM 25 N PHE A 4 39.649 2.772 13.343 1.00 10.60 N ATOM 26 CA PHE A 4 40.460 3.622 14.191 1.00 6.54 C ATOM 27 C PHE A 4 41.743 4.004 13.535 1.00 15.30 C ATOM 28 O PHE A 4 42.115 5.146 13.569 1.00 9.36 O ATOM 29 CB PHE A 4 40.730 2.949 15.557 1.00 11.91 C ATOM 30 CG PHE A 4 39.448 2.856 16.342 1.00 18.40 C ATOM 31 CD1 PHE A 4 38.580 1.770 16.196 1.00 7.82 C ATOM 32 CD2 PHE A 4 39.061 3.897 17.184 1.00 17.51 C ATOM 33 CE1 PHE A 4 37.402 1.658 16.933 1.00 20.71 C ATOM 34 CE2 PHE A 4 37.877 3.807 17.920 1.00 17.71 C ATOM 35 CZ PHE A 4 37.047 2.691 17.804 1.00 13.35 C |
Fortunately the reflections have been deposited for most of the TNT structures that have this problem, so in future PDB_REDO runs we are likely to see this problem get solved.
The other messages that WHAT_CHECK can produce should all be clear to crystallographers, and both meaningless and not very important for others.