Figure 1. Histogram of an example dataset that shows a nearly normal distribution.
Proteins are very simple molecules. They are linear chains of residues. And there are only 20 different residue types.
So far for the theory....
The practice is different:
These two points combined are the origin of "incorrect structures" and "weak points in generally correct structures".
A molecular biologist, working on a big project to elucidate the function of a certain protein, turns to somebody in a biocomputing group with a very specific question. They initiate a database search to find out what is known about the structure, and a very similar protein turns up from the Protein Data Bank. A dream comes true? Can important conclusions be deduced from this structure?
It is important to realize that any model based on the structure they found will be at least as bad as the structure!
It is obvious that structures that are very old might be less accurate, because refinement techniques 15 years ago were not so advanced. So, as a first indication, the year of publication can be used to see whether you might want to use the structure. But this criterion is not very convincing...
Somewhat more sophisticated is to look at the resolution of the X-ray structure. The lower the number, the more data the crystallographer had available, and the more chance that any mistake would have been detected. But the resolution just gives you an approximate indication of how well the structure can be theoretically. It doesn't tell you anything about which parts of the structure are good and which parts can not be trusted. It also doesn't warn you if there is a serious mistake somewhere.
A lot more sophisticated is to use a protein structure verification program like WHAT IF's "CHECK" menu. It will give you a detailed analysis of the protein, giving not only overall numbers but also specifying possibly problematic residues. And it uses a graphical representation of results to get a quick overview of the structure.
Crystallographers are refining a structure. They would like to know
how they're doing. Is the overall trace reasonable, or is there a shift
in this helix? Do all the geometries look as expected, or are some
side-chains in strange conformations? Is there a good alternative
conformation for the backbone in this loop?
Should they deposit the structure already?
The crystallographer or NMR spectroscopist can use WHAT_CHECK to check whether there are weak
points in his structure, using the results to improve it before deposition.
The big advantage is that he has the original X-ray data available, and is
still working on the refinement. To correct problems at a later stage would
be much more difficult.
In 1963, G. N. Ramachandran, C. Ramakrishnan and V. Sasisekharan wrote a paper about a graphical representation of the two most important backbone torsion angles (phi and psi) (J.Mol.Biol 7:95-99 (1963)). They presented a simple theoretical model, showing that in a phi/psi plot all residues are fairly restricted in their possibilities.
This φ,ψ plot, later called "Ramachandran plot", was the first serious verification tool for protein structures. Structures that were solved before 1963 were solved without knowledge of Ramachandran's work, and thus the Ramachandran plot can be used as an independent judgment of these structures. In later structures (after 1963), the crystallographers could be aware of the work, and could use it to help during the refinement. So, in principle, there should not be any structures with bad Ramachandran plots that have been deposited after 1963.
However.... There are two reasons why there are still structures with strange looking Ramachandran plots deposited in the Protein Data Bank much later than 1963:
The last point in the previous paragraph brings up a nice point: of course it is good if a verification tool can be used during refinement. If such a tool is widely used, it will improve the quality of the new structures, and probably make structure solution easier (more automatic) at the same time.
But it is also nice if some of the verification tools can not be used during the refinement. These tools can later be used for an "independent" judgement of the structure. A number of such checks are part of WHAT IF's protein structure verification menu.
Protein verification checks work by comparing structural parameters to standard values. These standard values can be obtained in different ways:
Almost any number one can use to describe a structure has some uncertainty. For instance a single-bond distance between 2 carbon atoms in a small molecule structure is about 1.53 Angstroms, but some are a bit longer, and some are a bit shorter. Say: most C-C bonds in small molecule structures are between 1.50 and 1.56 Angstroms long.
Where does this variation come from?
When considering the variation observed in a certain variable, it is of utmost importance to judge the influence of the two mentioned effects on the variability. Looking at the example of a small molecule C-C bond distance, the natural variation is much larger than the inaccuracy, and thus the variation observed ("A normal C-C bond length is between 1.50 and 1.56 Angstrom") is relevant.
For a C-C bond in an unrestrained protein structure, the opposite would hold. The natural variation will be lower than for small molecules, because the variability in local connectivity is much smaller. The inaccuracy in the determination of the bond length, however, is much larger than for small molecule structures. The combination of these two differences makes the 2nd effect much larger than the 1st, making the observed variation ("A C-C bond length in a protein is between 1.1 and 2.0 Angstrom") absolutely irrelevant for the understanding of protein structures.
Lets assume we have found a parameter that is worth studying: we have 1000 C-C distances from reliable small molecule structures. Any standard statistics package will tell you something like "the average is 1.532, the standard deviation of the population is 0.021".
What does this tell us about a structure with a 1.66A C-C bond?
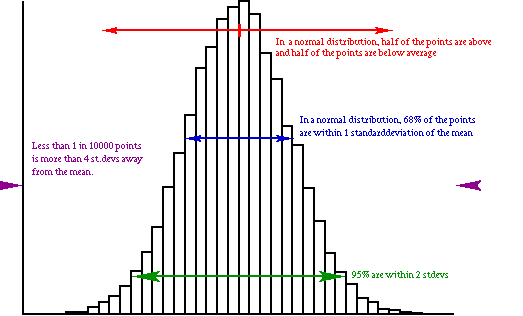
Lets take a look at a simulated normal distribution:
|
|
Figure 1. Histogram of an example dataset that shows a nearly normal distribution. |
From this picture it is clear that:
Coming back to our 1.66A bond, what can we say? The distance to the mean is (1.66-1.532)=0.128 Angstrom, which is about 6 standard deviations. This is highly unlikely, and would definitely warrant further study!!
Now lets do something similar for a protein. Lets say we have a 400 residue protein. For each of the bonds in this protein (approximately 4000) we do an analysis like the one above. Now we find 1 that is 4.5 standard deviations away from the mean, all others are less than 4.0. Is this one bond length deviation an error? Not really, because we expect 1 in 10000 to be more than 4.0 standard deviations away from the mean, and we studied 4000 numbers. One deviation seems to be allowed here. On the other hand, what makes this one bond so special that it wants to deviate more than all the other ones? This indicates a fundamental feature of protein structure verification: It is completely normal to find a few outliers, but it is always worth investigating them. But: if outliers are not exceedingly rare, there is something strange going on....
You might have noticed that we need the phrase "standard deviations away from the mean" quite a lot. Mathematicians hate repeating long phrases, and they have given this a new name: The number of "standard deviations away from the mean" is called "Z". Formally: Z is the measured value minus the "mean", divided by the "standard deviation of the population", or:
X - mu
Z = --------
sigma
|
So Z is negative if the value "X" is less than the mean, and Z is positive if the value is greater than the mean. "Outliers" now are all values with Z<-4 or Z>4. WHAT IF uses this criterion a lot to decide which values need to be listed.
Something else: Z has a very nice property for doing statistics. This can help us to judge whether outliers are indeed rare, or whether there are more (or less!) outliers than expected. This property is: The "root mean square" of a population of Z values should be 1.0. So for our hypothetical 400 residue protein:
,-----------------
\ / sum (Z2)
RMS-Z = \ / ---------------
\/ number of bonds
|
should be approximately 1.0. WHAT IF contains a number of these tests, and will complain if any of these values deviates from 1.0 in an "abnormal" way. This is normally a very sensitive indicator!
Summarizing: There is a "Z-score" and an "RMS-Z-score". A Z-score should be 0, and most of the time if it is negative it means worse than average, and positive better than average. An RMS Z-score should be close to 1.0. Sometimes any deviation from 1.0 is "bad" (e.g. bond distances), in other cases one direction is "good" and the other is "bad". WHAT IF will give a "subjective" annotation to indicate whether a value is "good" or "bad".