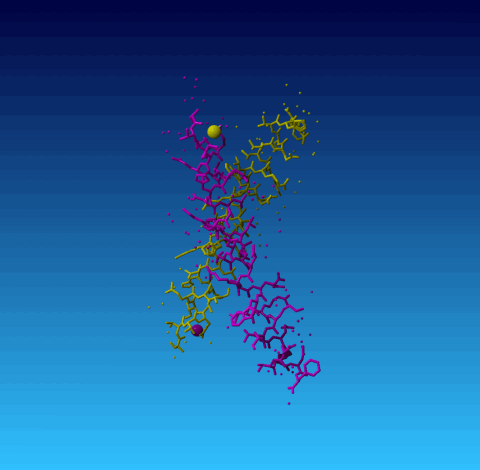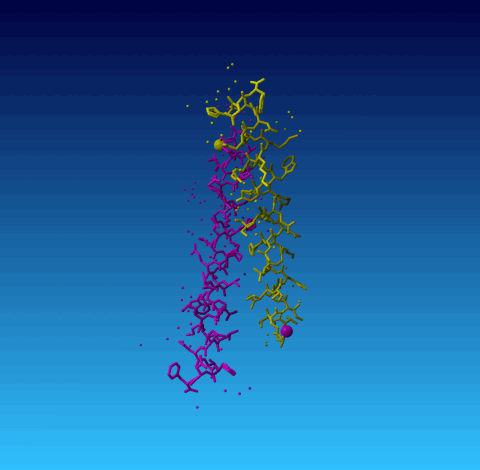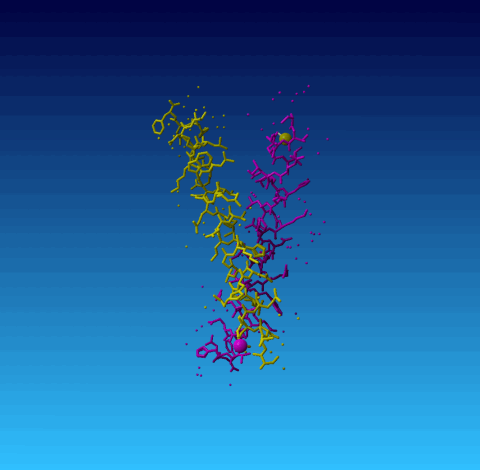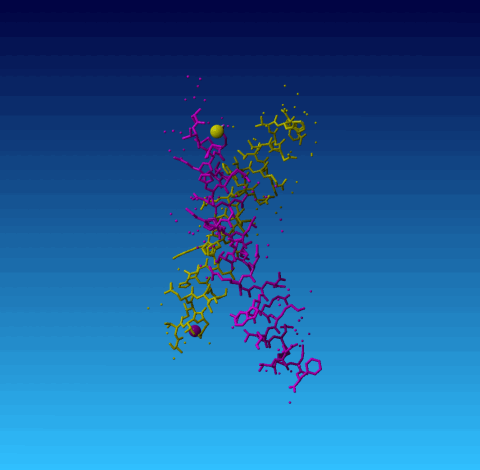
Figure 2. 1ET1 Everything with chain identifier A is in yellow, and everything with chain identifier B in purple. The small balls are water molecules. The two big balls are sodium ions.
The WHAT IF program uses the famous 'SHOSOU' command to analyze the contents of a PDB entry. Inside WHAT IF / WHAT_CHECK this content is called 'the SOUP', because after all a PDB file and a cup of soup both consist of water with proteins in it. The only differences are the order and the taste.
A typical result from the extended SHOSOU command looks like:
Contents of the SOUP: *1
Protein .................... : 2 *2
Drug, ligand or co-factor .. : 1
DNA or RNA ................. : 0
Single atom entity ......... : 7
(Groups of) water .......... : 1
Drug with known topology ... : 0
Molecule Range Type Set name *3
1 1 ( 1) 316 ( 316)E Protein set *4
2 317 ( 322) 318 ( 323)D Protein set *4
3 319 ( O2 ) 319 ( O2 )E K O2 <- set *5
4 320 ( 317) 320 ( 317) CA set *6
5 321 ( 318) 321 ( 318) CA set
6 322 ( 319) 322 ( 319) CA set
7 323 ( 320) 323 ( 320) CA set
8 324 ( 321) 324 ( 321) ZN set
9 325 ( 324) 325 ( 324) DMS set *7
10 326 ( O2 ) 326 ( O2 )D L O2 <- set *8
11 327 ( HOH ) 327 ( HOH ) water ( 157) set *9
*10 *11 *12 *13 *14 *15 *16
|
After showing the content of the PDB file (which in WHAT IF / WHAT_CHECK terms is 'the SOUP') you get some countings, like the number of residues, the number of waters, and the numbers of those that have unlikely or missing atoms. WHAT_CHECK also looks for residues with a negative (or zero) residue number, and it looks for consecutive residues with decreasing residue numbers.
In this section you also find some statistics about the use of chain identifiers. There is nothing wrong is a series of molecules have as chain identifier A,B,C,E,F,G, respectively. But the missing chain C might be indicative for an administrative problem that the experimentalist might immediately recognize.
This list is, just like the SHOSOU table more meant for the experimentalist who might see something in his/her PDB file that isn't supposed to be there.
In case ions are found that have the wrong chain identifier, they are listed in a table. An ion is said to have the wrong chain identifier if its chain identifier is the same as that of a protein, nucleic acid, or sugar chain, while it makes more contacts with a protein, nucleic acid, or sugar chain with another chain identifier. Obviously, this isn't wrong, but is surely doesn't help the end-users. An example is found in 1ET1:
JRNL AUTH L.JIN,S.L.BRIGGS,S.CHANDRASEKHAR,N.Y.CHIRGADZE, JRNL AUTH 2 D.K.CLAWSON,R.W.SCHEVITZ,D.L.SMILEY,A.H.TASHJIAN, JRNL AUTH 3 F.ZHANG JRNL TITL CRYSTAL STRUCTURE OF HUMAN PARATHYROID HORMONE JRNL TITL 2 1-34 AT 0.9-A RESOLUTION. JRNL REF J.BIOL.CHEM. V. 275 27238 2000 |
WHAT_CHECK reports for this file:
# 22 # Warning: Ions bound to the wrong chain ============================================= The ions listed in the table have a chain identifier that is the same as one of the protein, nucleic acid, or sugar chains. However, the ion seems bound to protein, nucleic acid, or sugar, with another chain identifier. Obviously, this is not wrong, but it is confusing for users of this PDB file. 71 NA ( 101-) A - 72 NA ( 102-) B - |
|
|
Figure 2. 1ET1 Everything with chain identifier A is in yellow, and everything with chain identifier B in purple. The small balls are water molecules. The two big balls are sodium ions. |
In the box below we illustrate the residue and atom nomenclature with one warning as an example.
Warning: Unusual bond angles The bond angles listed in the table below were found to deviate more than 4 sigma from standard bond angles (both standard values and sigma for protein residues have been taken from Engh and Huber [REF], for DNA/RNA from Parkinson et al [REF]). In the table below for each strange angle the bond angle and the number of standard deviations it differs from the standard values is given. Please note that disulphide bridges are neglected. Atoms starting with "-" belong to the previous residue in the sequence. 1 THR ( 2-) A - CA CB OG1 103.06 -4.4 1 THR ( 2-) A - CG2 CB OG1 117.41 4.1 12 ASN ( 13-) A - ND2 CG OD1 127.61 5.0 14 ASN ( 15-) A - ND2 CG OD1 128.63 6.0 39 THR ( 40-) B - CA CB OG1 103.59 -4.0 45 ALA ( 46-) B - N CA CB 103.98 -4.3 *1 *2 *3 *4 *5 *6 *6 *6 *7 *8 |
The box lists a warning. A series of bond-angles considered unusual is listed. The *1, *2, etc is not part of the output but added here to label the columns.