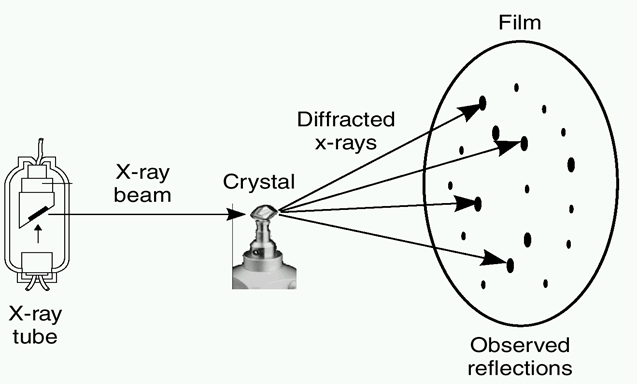
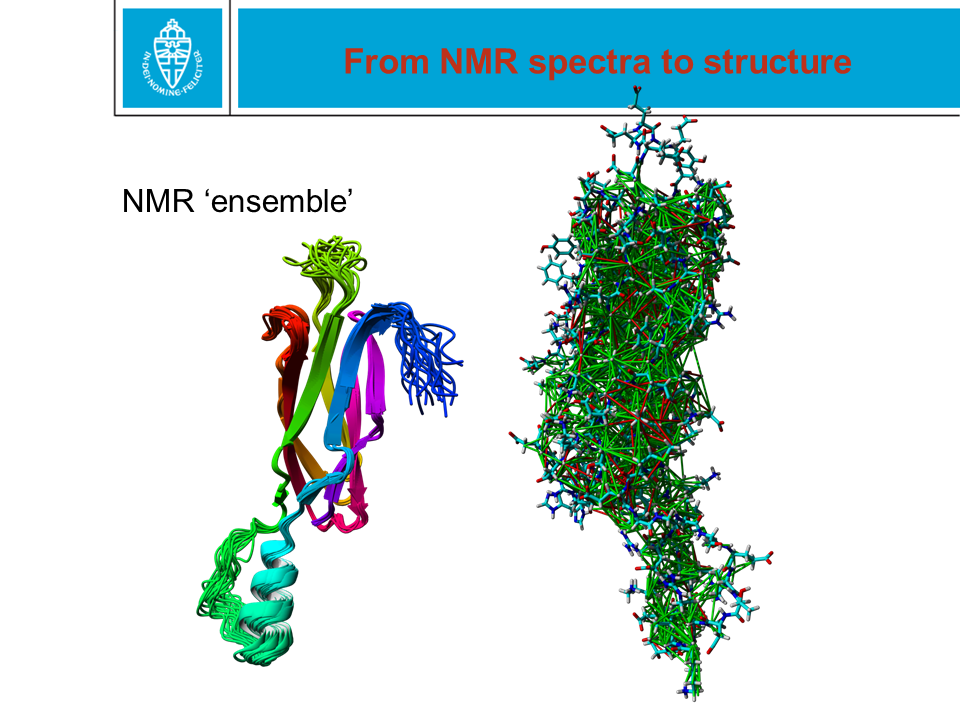
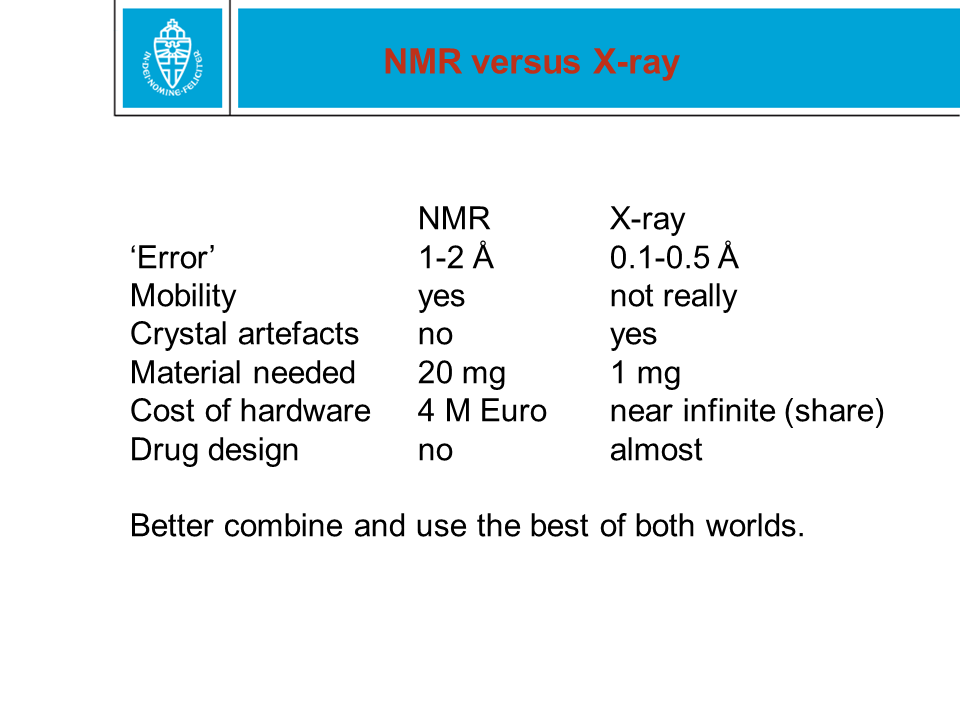
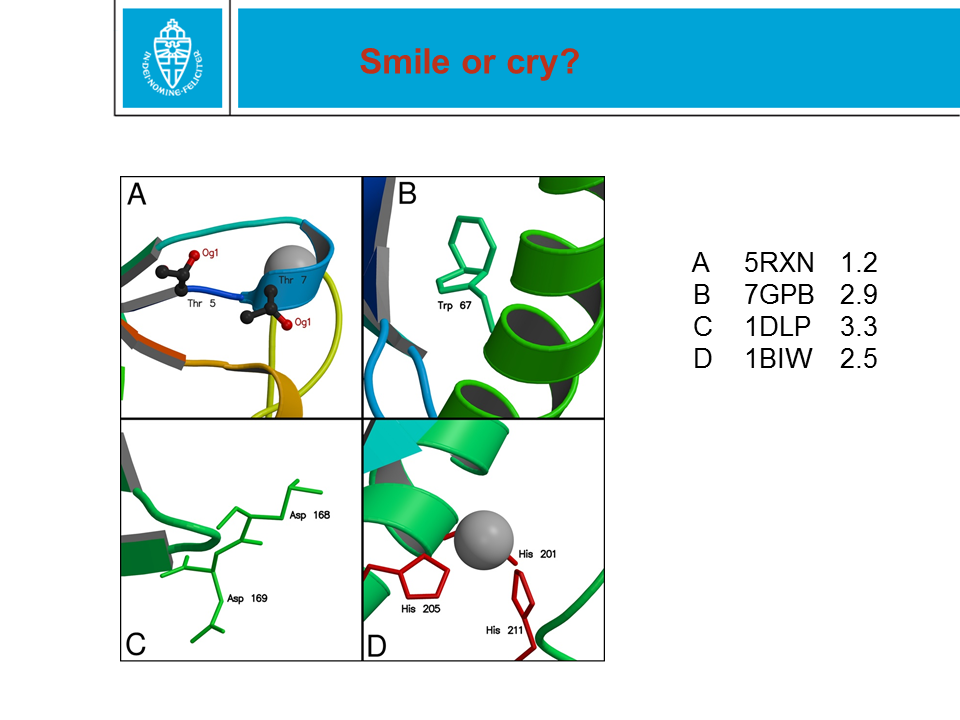
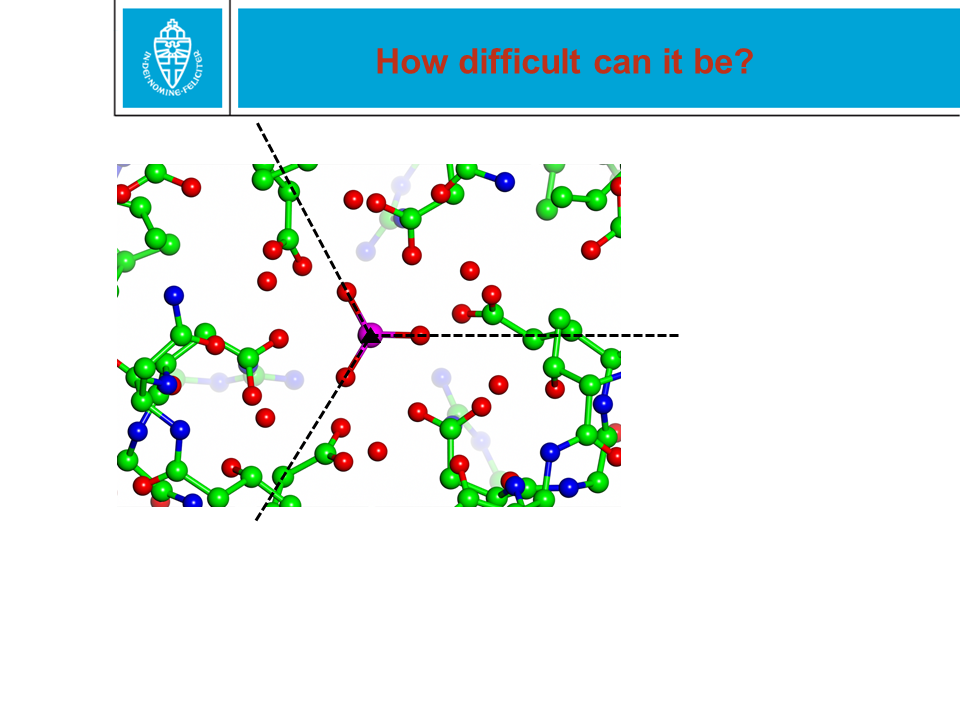
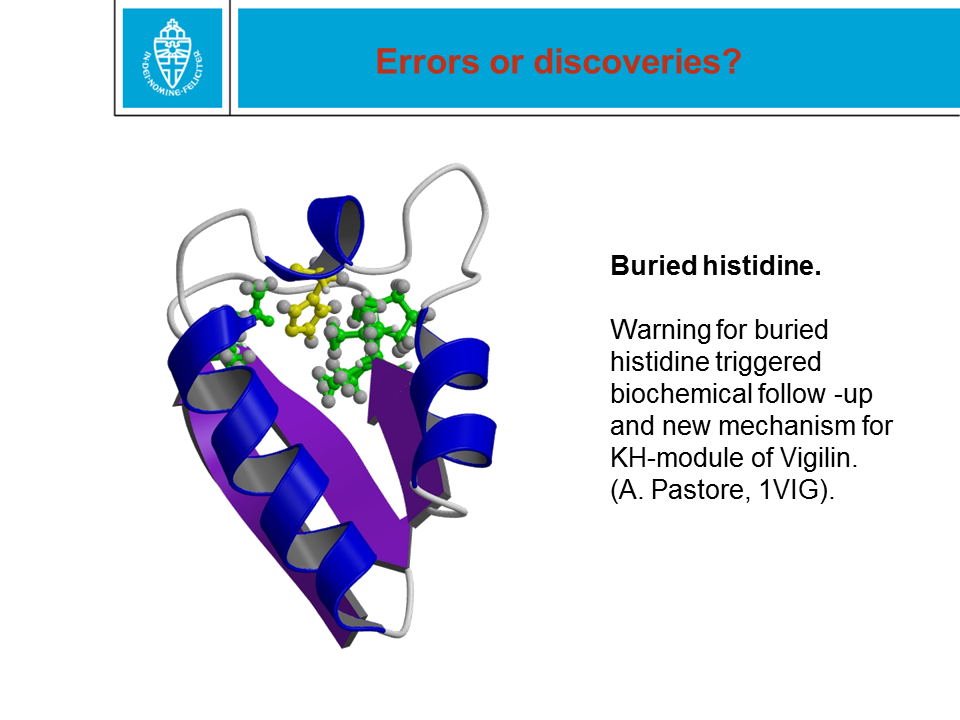
Figure 28. When a protein structure is being solved by X-ray or NMR,
coordinates are obtained for the (most) atoms. These two methods are
technologically and scientifically very different. In the PDB file,
coordinates are given with an error margin of only 0.0005 Ångström. That
precision is NOT realistic.
![]()