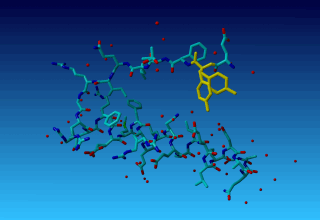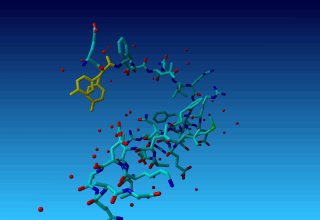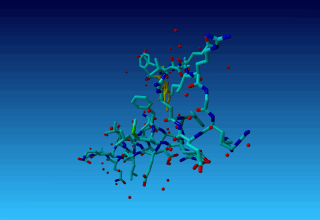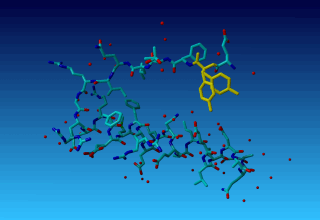
Figure 36. Tyrosine 327 in this structure you see that the (yellow) tyrosine has two side chains.
|
In this tutorial we will show you: |
Read the exercise file 1AIE.pdb in YASARA.
|
|
Figure 36. Tyrosine 327 in this structure you see that the (yellow) tyrosine has two side chains. |
Question 18: Why does the second residue in this tiny molecule already has residue number 327? Can you explain this seemingly Belgian numbering scheme?
Answer
Question 19: Why does TYR-327 have two side-chains? (We will ask in the docking practical how this two-headed hydra-behaviour can help docking software determine which residues have a flexible side chain).
Answer
Now load the file 1ppt_crystal.sce from the Exercise files (it is called a big sphere...). In this scene you see a sphere that I cut out of the crystal they used to solve the structure of 1ppt. Obviously this scene holds so very many atoms that you will never recognize anything (except that I drew the one central copy of 1ppt in ball-mode. Study this packing .
Question 20: Is there space for much water in the crystal of 1ppt?
AnswerOpen the PDBREPORT for 1AIE in another window.
Question 21:
According to the PDBREPORT, which residue in 1aie should have
its side chain flipped for
optimal hydrogen bonding
What does it actually mean: "Error: HIS, ASN, GLN side chain
flips"?
Look at the structure. Do you understand why WHAT_CHECK claims that this
residue should be flipped? If you have no idea, then get the exercise
file 1AIE that is already 'flipped' for you, and see which H-bonds you
now find extra.
Get the file 1PPT.pdb in YASARA, and get the PDBREPORT for the PDB file 1ppt in another window.

|
Figure 37. The structure of 1ppt contains a Zn atom. That is weird, because 1ppt is not an enzyme, and a zinc that is only bound by two non-charged atoms is not likely to significantly contribute to the protein's stability. |
Question 22: Why is that zinc there?
Answer
Question 23:
|
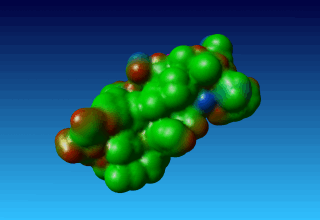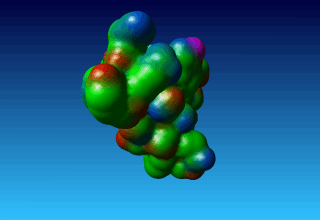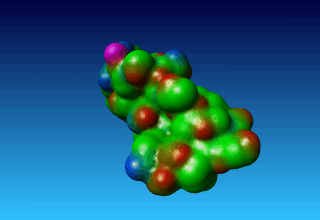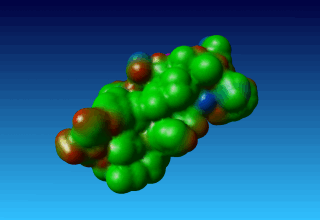
Figure 38. the surface of 1ppt coloured by atom type (carbon=green, oxygen=red, nitrogen=blue). |
Question 24: In the PDB report for 1ppt I find:
Sect 8) Packing, accessibility and threading Warning: Inside/Outside residue distribution unusual The distribution of residue types over the inside and the outside of the protein is unusual. Normal values for the RMS Z-score below are between 0.84 and 1.16. The fact that it is higher in this structure could be caused by transmembrane helices, by the fact that it is part of a multimeric active unit, or by mistraced segments in the density. |
When you look at 1ppt, you might see something particular about the distribution of hydrophobic and hydrophilic residues (if you load the YASARA scene 1ppt_foobfiel.sce from the exercise files in YASARA, you will find the the hydrophobic residues coloured greenish and green, the hydrophilic ones are red, and the inbetween ones are orange/brown.
Normally you expect the surface of a protein to have hydrophilic residues more or less spread over the surface, but for 1PPT we see something different.
Question 25:
Think of a reason for the weird distribution of hydrophobic and hydrophilic
residues. Feel free to be inspired by the WHAT_CHECK warning!
Hint 1: What does the word hydrophobic mean?
Hint 2: What does the word multimer mean?
Answer
Question 26:
WHAT_CHECK, correctly, warned for a weird inside/outside distribution of
hydrophobic and hydrophilic residues. It gave this
distribution a score and listed the score-limits for normal proteins. So,
obviously, some kind of force field must be involved. In no more
than 20 lines, describe how the WHAT_CHECK authors produced this force field,
and how they determined the limits for normally distributed proteins.
Hints: 1) Think about the data you can obtain from the PDB; 2) Think how
these data can be represented numerically; 3) Think how you can quantify
those numerical data with one form of statistics or the other; 4) Think of
a simple algorithm (computer program) that can use the resulting data to score
your protein.
We will discuss one more aspect of protein structure validation. In the report you see two rather bad errors and many, many small errors. The one bad error is a 12 sigma planarity problem, and the other bad error is a couple of 'flipped' residues.
Find these problems in the WHAT_CHECK report, and find them in the gvbc.pdb PDB file.
Question 27: What kind of molecule(s) do you see in the active site of the gvbc enzyme?
Answer
Question 28: Which of the two aforementioned errors is worse for the ligand binding?
Answer
Question 29:
So, draw a conclusion about these last two questions. Why is it important to
report a 12σ planarity deviations if the next thing I tell you is
that 'it is just a minor scratch' to quote Mothy Python in one of my favourite
movies? And on the other hand, if I flip an asparagine, then that does nothing
to the R-factor or the fit of the atoms to the density,
and still I call it a very serious problem in this structure. Why?
Feel free to discuss this typical exam question with the assistant(s) before
you go home.