Practical 1
What is a force-field? Well, there are many things one can call a force-field,
but in protein structure related bioinformatics it normally tends to be a
dataset derived from known protein structures (or peptidic small molecules)
combined with a set of rules that together can be used to 'score' a protein
or an aspect of a protein, or that together can be used to predict something
about a protein.
In this practical, for example, we are going to 'count' residue types in
transmembrane helices and from the results we are going to make a force field
that can be used either to see how happy known transmembrane helices are, or
to predict transmembrane helices in a protein for which no structure information
is available.
Bond lengths
One of the simplest force-fields you have encountered so far has been the
validation of bond lengths. Ideal bond lengths were determined twenty years
ago by Engh and Huber. They found values
like δ(N,Cα)= 1.458 +/- 0.019.
Question 30:
Engh and Huber (EH) used the CSD to determine the ideal bond lengths. And in
the validation seminar we called the number of standard deviations that any
bond length deviates from the ideal value its Z-score. Structure validation
software normally complaints about bonds for which that Z-score is > 4.0.
value
What is the CSD?
Why did Engh and Huber use the CSD?
Why is the ideal δ(N,Cα)= 1.458 +/- 0.019 and not
simply 1.458 +/- 0.0?
Answer
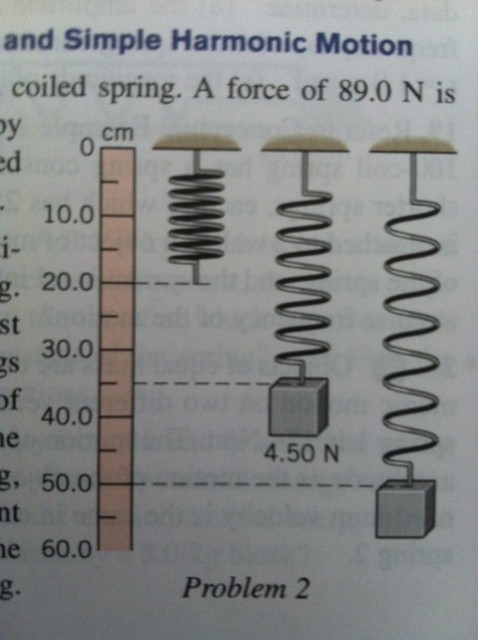
|
Figure 48. We have already two times looked at the formula for bond legths
that we will discuss in the MD seminar:
V = 1/2 K * (b-b0)2. This
formula looks very much like the energy as function of stretching a spring.
With that in mind, can you see the similarity in the terms in the equations
used for the weight hanging at a spring and the equation for the bond length b
in this MD equation for V (the potential energy of the bond at length b)?
|
Question 31:
How are the Engh and Huber bond lengths and standard deviations used
in Molecular Dynamics packages?
Answer
Question 32:
Last week you have seen that the Engh and Huber bond lengths are used in
structure validation. Look at the PDBREPORT for 1PPT and find out how and where
the bond lenghts are used in the report.
Answer
Question 33:
Engh and Huber also determined the ideal bond angles. In analogy to the bond length
parameters and force field discussed above, describe:
How did Engh and Huber determine the parameters underlying the bond angle force field?
Which formulas did Engh and Huber use to convert the bond angle data into a force field?
Describe what the Engh and Huber bond angle force field looks like?
Describe how WHAT_CHECK uses the Engh and Huber bond angle force field for structure validation.
Answer
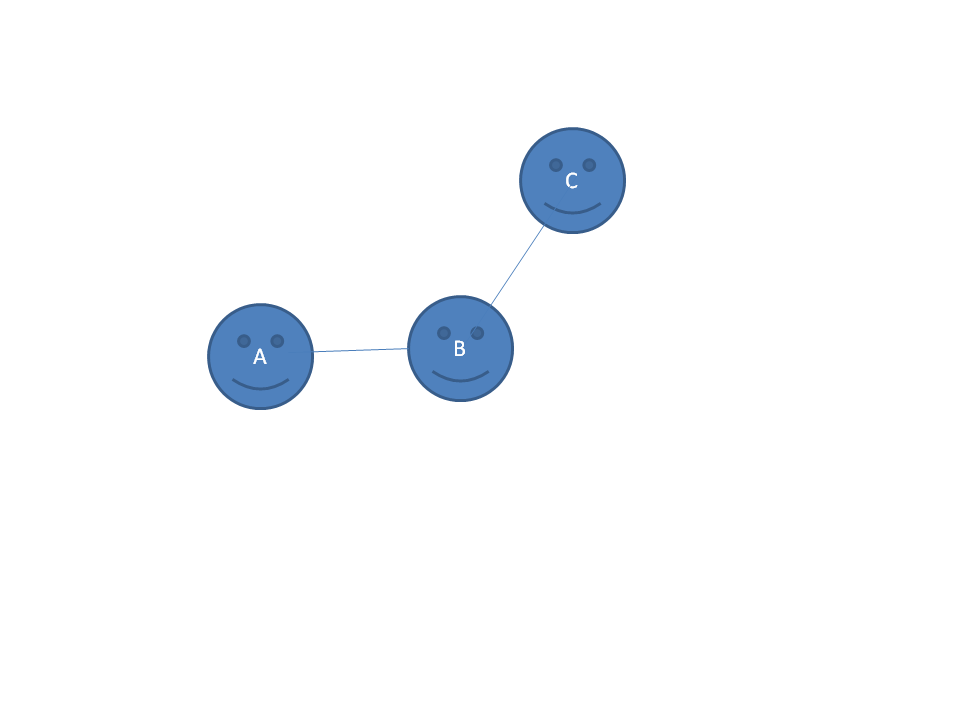
Making a baby-FF
Lets look at the small molecule A-B-C. We have 10 molecules in the CSD database
that contain the A-B-C sub-structure as a fragment. It is known that B
is SP2 hybridized. This molecule contains a bunch of protons, but those are
too small to be seen anyway, so we forget all about them for today.
We measured the ten A-B and B-C bond lengths and found:
A-B distances: 1.45,1.47,1.44,1.46,1.49,1.46,1.45,1.44,1.47,1.46
B-C distances: 1.52,1.59,1.46,1.50,1.55,1.62,1.55,1.51,1.49,1.60
|
Question 34:
What can you say about the lengths of these two bonds?
(Hint: Look here.)
Answer
|
Figure 49. I have solved the structure of A-B-C, and in my structure I find that the
distance A-B is 1.519 and the distance B-C is 1.630
Now get a paper
copy of this plot from the assistants, and write the numbers you determined,
and the numbers I observed, and your conclusions from the next question
in this plot.
|
Question 35:
In my structure both distances are a bit wrong. How wrong? And which of the
two is "wrongest"?
Answer
Now we are going to energy minimize the atoms in my A-B-C molecule. This exercise
is a prelude to the molecular dynamics seminar. Actually, we are not going to minimize
the energy, but we are going to figure out the concepts needed to do it, if one
day we had to...
Question 36:
Design an algorithm that uses all data collected in this A-B-C project to improve
the structure. Discuss the algorithm with the assistants before continuing.
Answer
After discussing the plan with the assistants, take the four forces, convert them
into 3 vectors, and draw where the atom centres will end up after you move them
according to the vector direction and magnitude.
Look at the new plot and and discuss what happened.
It is very likely that the structure at the end is not yet very good. Certainly
the A-B-C angle is no longer 120 degrees. And funny enough, if you gave the vectors
exactly the right length to bring the two distances in order, then after applying
both vectors to B, both distances stayed too long.
Question 37:
Summarise what we did. Summarise what we did wrong. Think of how to do
it better. And if you cannot come up with a good answer, then wait for a while
and listen to the MD seminar.
Answer
Protein membrane interactions
Lets design a force-field to predict whether a protein goes through the membrane with
one or more helices. We do this in two steps:
- Going through a thinking experiment to see which steps are needed
- Making a real functioning force field
Use the series of questions as guide to come up with a detailed plan for the implementation
of this FF (and remember Google is your friend).
Question 38:
How many residues are needed to stick a helix through a membrane?
Answer
Question 39:
What should your force-field decide? How many states should it recognize?
(Do not continue before you have discussed this topic with the assistant(s)).
Answer
Question 40:
What do you need as data?
Answer
Question 41:
What are you going to count in the FF data collection stage?
Answer
Question 42:
What will be your null-model?
(Discuss this point with the assistants before you continue).
Answer
Question 43:
Now, lets think a bit about the amino acids. Which amino acids do you expect to
observe more often in the TM helices? And which much less?
Answer
Question 44:
And how do we now get numbers that we can do something with? Obviously we want to get
some TM-score for each residue type.
Answer
Question 45:
How are you now going to predict TM helices?
Answer
Take a look at the GPCRDB. This database (www.gpcr.org/7tm/) holds, among other things,
multiple sequence alignments of GPCRs, and those are molecules with 7 transmebrane helices.
Go to 'browse families', to the 'Hormone protein' family, and to 'Mview alignment'. This
is a big file, so just remain calm and wait.
Question 46:
Summarize the residue colouring scheme in one short sentence.
Can you recognize the seven helices in the alignment?
Funny enough about each of the 7 helices contains at least one polar residue. Where do you expect
that these polar residues are located in the structure, inside or outside?
Answer
And now for the real thing.
Lets make a force field to predict transmembrane helices
in G protein-coupled receptors (GPCRs).
To do so we need data to set-up the force field. Normally, we would take a large set of membrane
proteins with know structure, but as this practical is only meant to illustrate the concepts,
we will use just one protein to generate the data.
Today we will not only design and implement a force field to predict transmembrane
helices in GPCRs, but we will also test our force field. To achieve this double goal,
we will use data only from one GPCR to set-up the force field, and we
use a totally different GPCR to see how well the force field works.
But first a short intro in GPCRs:

|
Figure 50. The structure of opsin; the first GPCR for which the structure was
solved. Nearly all GPCRs have a series of residues in common. The side chains
of these conserved residues are shown in this figure. These conserved residues
allow us to align GPCR sequences very easily. In the exercise files you
find the YASARA scene opsin.sce. This scene produced this figure, and in this
scene all residues have the same numbers as in the GPCRDB.
|
In the box below, you find the secondary structure determination of the
opsin file shown in figure 50. In this box the conserved residues are numbered
above the sequence using the GPCRDB-numbering scheme for easy reference in case you
want to look things up in the GPCRDB.
130
|
1 - 60 MNGTEGPNFYVPFSNKTGVVRSPFEAPQYYLAEPWQFSMLAAYMFLLIMLGFPINFLTLY
1 - 60 TSS TT SSTT TTT TTTT 333T HHHHHHHHHHHHHHHHHHHHHHHHHHH
220 224 305
| | |
61 - 120 VTVQHKKLRTPLNYILLNLAVADLFMVFGGFTTTLYTSLHGYFVFGPTGCNLEGFFATLG
61 - 120 HHHHTTT THHHHHHHHHHHHHHHHHHHTHHHHHHHHHHTT TTHHHHHHHHHHHHHHH
340 420
| |
121 - 180 GEIALWSLVVLAIERYVVVCKPMSNFRFGENHAIMGVAFTWVMALACAAPPLVGWSRYIP
121 - 180 HHHHHHHHHHHHHHHHHHHT TTT HHHHHHHHHHHHHHHHHHHTHHHHTTT SSS
520 528
| |
181 - 235 EGMQCSCGIDYYTPHEETNNESFVIYMFVVHFIIPLIVIFFCYGQLVFTVKEAAA
181 - 235 STTTTSSSS T TTTTHHHHHHHHHHHTTHHHHHHHHHHHHHTTTT T
620
|
236 - 295 SATTQKAEKEVTRMVIIMVIAFLICWLPYAGVAFYIFTHQGSDFGPIFMTIPAFFAKTSA
236 - 295 TTTTHHHHHHHHHTHHHHHHHHHHHHHHHHHHHHHHTTT HHHHHHHHHHHHHHH
730
|
296 - 323 VYNPVIYIMMNKQFRNCMVTTLCCGKNPSTTVSKTETSQVAPA
296 - 323 HHHHHHHHHH HHHHHHHHHHHTTT TTTTT
|
Question 47:
Do the helices that you see with YASARA, and the helices indicated in the box agree?
And if they don't, why?
Answer
The GPCR force field
And now the important part. Work in groups of three or four students, and
come up with a plan for the force field design.
(hint: the remainder of this paragraph holds useful information...)
Let your design be guided by:
Question 48:
1) Which data do you need?
2) What will be your null-model? In other words, which two things are you
going to compare?
3) How are you going to get the data?
4) How are you going to make the counting easy? (We are not going
to manually count the thousands of residues we need to get a reliable
answer).
5) Which formula are you going to use to convert the data into something
that can be used predictive?
6) What should the software look like that does the prediction in the end?
Answer
All the counting has already been done for you, albeit that the counting results
have a somewhat clumsy format: the HSSP format. HSSP files consist of a header:
HSSP HOMOLOGY DERIVED SECONDARY STRUCTURE OF PROTEINS , VERSION 1.1 2001
PDBID 1f88
DATE file generated on 17-May-10
SEQBASE UniProtKB/Swiss-Prot(20-Apr-2010) UniProtKB/TrEMBL(20-Apr-2010)
PARAMETER SMIN: -0.5 SMAX: 1.0
PARAMETER gap-open: 3.0 gap-elongation: 0.1
PARAMETER conservation weights: YES
PARAMETER InDels in secondary structure allowed: YES
PARAMETER alignments sorted according to :DISTANCE
THRESHOLD according to: t(L)=(290.15 * L ** -0.562) + 5
REFERENCE Sander C., Schneider R. : Database of homology-derived protein structures. Proteins, 9:56-68 (1991).
CONTACT Maintained at www.CMBI.ru.nl by Elmar.Krieger.cmbi.umcn.nl
AVAILABLE Free academic use. Commercial users must apply for license.
Etcetera.
|
The section that lists all aligned sequences:
## PROTEINS : EMBL/SWISSPROT identifier and alignment statistics
NR. ID STRID %IDE %WSIM IFIR ILAS JFIR JLAS LALI NGAP LGAP LSEQ2 ACCNUM PROTEIN
1 : OPSD_BOVIN 3DQB 1.00 1.00 565 648 243 326 84 0 0 348 P02699 RecName: Full=Rhodopsin;
2 : OPSD_BOVIN 3DQB 1.00 1.00 342 483 1 142 142 0 0 348 P02699 RecName: Full=Rhodopsin;
3 : OPSD_BOVIN 3DQB 1.00 1.00 1 235 1 235 235 0 0 348 P02699 RecName: Full=Rhodopsin;
4 : OPSD_BOVIN 3DQB 1.00 1.00 237 324 240 327 88 0 0 348 P02699 RecName: Full=Rhodopsin;
5 : OPSD_BOVIN 3DQB 1.00 1.00 485 563 148 226 79 0 0 348 P02699 RecName: Full=Rhodopsin;
6 : OPSD_SHEEP 0.99 1.00 342 483 1 142 142 0 0 348 P02700 RecName: Full=Rhodopsin;
7 : OPSD_FELCA 0.99 0.99 342 483 1 142 142 0 0 348 Q95KU1 RecName: Full=Rhodopsin;
8 : OPSD_PHOGR 0.99 0.99 342 483 1 142 142 0 0 348 O62795 RecName: Full=Rhodopsin;
9 : OPSD_PHOVI 0.99 0.99 342 483 1 142 142 0 0 348 O62794 RecName: Full=Rhodopsin;
10 : Q2KND7_URSMA 0.98 0.99 358 483 1 126 126 0 0 322 Q2KND7 SubName: Full=Rhodopsin;
11 : Q2KND6_PHOHI 0.98 0.99 358 483 1 126 126 0 0 324 Q2KND6 SubName: Full=Rhodopsin;
12 : OPSD_CANFA 0.98 0.99 342 483 1 142 142 0 0 348 P32308 RecName: Full=Rhodopsin;
Etcetera
|
The alignment part is important to find the residue numbers back for the helices (I coloured the
secondary structure assignments red; a H in this column, obviously, indicates a helix).
## ALIGNMENTS 1 - 70
SeqNo PDBNo AA STRUCTURE BP1 BP2 ACC NOCC VAR ....:....1....:....2....:....3....:....4....:....5....:....6....:....7
1 1 A M 0 0 192 292 3 M M M MMMMM M MM MMM M MMMM
2 2 A N + 0 0 72 430 27 N N N NNNNN N NN NNN N NNNN
3 3 A G S S- 0 0 5 431 33 G G G GGGGG G GG GGG G GGGG
4 4 A T E -A 11 0A 28 453 23 T T T TTTTT T TT TTT T TTTT
5 5 A E E -A 10 0A 112 465 21 E E E EEEEE E EE EEE E EEEE
6 6 A G - 0 0 10 474 21 G G G GGGGG G GG GGG G GGGG
7 7 A P S S+ 0 0 108 466 51 P P P PPPPP P PP PPP P PPPP
8 8 A N S S+ 0 0 72 493 51 N N N NNNNN N NN NNN N DNNN
9 9 A F - 0 0 18 503 17 F F F FFFFF F FF FFF F FFFF
10 10 A Y E -A 5 0A 10 518 39 Y Y Y YYYYY Y YY YYY Y YYYY
11 11 A V E -A 4 0A 0 547 32 V V V VVVVV V VVV VVV V IVVV
12 12 A P H S+ 0 0 6 525 33 P P P PPPPP P PPP PPP P PPPP
13 13 A F H S- 0 0 61 605 34 F F F FFFFF F FFF FFF F MFFF
14 14 A S H - 0 0 45 605 42 S S S SSSSS S SSS SSS S SSSS
15 15 A N H > + 0 0 68 609 20 N N N NNNNN N NNNHHNNN N NNNN
|
And the last section holds the profile (see the sequence alignment seminar and practical) from which you can get
an idea about the counting statistics.
## SEQUENCE PROFILE AND ENTROPY
SeqNo PDBNo V L I M F W Y G A P S T C H R K Q E N D NOCC NDEL NINS ENTROPY RELENT WEIGHT
1 1 A 3 2 0 95 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 292 0 0 0.234 8 0.94
2 2 A 0 0 0 0 0 0 0 0 0 0 5 0 0 0 3 0 1 11 65 15 430 0 0 1.153 38 0.47
3 3 A 0 10 0 1 0 0 0 69 0 0 14 0 0 0 0 3 0 0 0 0 431 0 0 1.057 35 0.36
4 4 A 0 0 0 3 0 0 0 0 0 9 1 78 0 0 0 0 2 4 0 2 453 0 0 0.912 30 0.56
5 5 A 0 1 0 0 0 0 0 0 0 1 3 0 0 0 5 0 10 78 0 1 465 0 0 0.890 30 0.62
6 6 A 0 0 0 0 0 0 0 70 3 2 7 0 0 0 0 0 0 7 0 11 474 0 0 1.062 35 0.56
7 7 A 0 1 2 0 13 0 0 0 0 34 17 1 0 0 0 14 2 9 4 3 466 17 1 1.943 65 0.10
8 8 A 0 0 10 0 13 7 10 0 5 0 0 0 0 0 0 5 0 5 33 11 493 17 0 2.036 68 0.10
9 9 A 0 1 10 1 80 1 1 0 0 0 0 0 0 4 0 0 0 1 0 1 503 17 0 0.817 27 0.65
Etcetera.
|
Obviously, we would like to have the counts for each residue type inside the helices and
inbetween the helices. Unfortunately, the profile gives you alignment occupancies
and percentages per residue type. The Web server called teller
can be used to convert the HSSP profile (called 1f88.hssp in the exercise files)
values into raw amino acid counts. Just cut-n-paste the values listed in the box
into the 'teller' software, and check that you get (and understand):
| aminozuur | aantal |
| A | 39 |
| C | 0 |
| D | 204 |
| E | 533 |
| F | 527 |
| G | 629 |
| H | 20 |
| I | 109 |
| K | 103 |
| L | 63 |
| M | 300 |
| N | 461 |
| P | 213 |
| Q | 69 |
| R | 36 |
| S | 213 |
| T | 358 |
| V | 9 |
| W | 40 |
| Y | 54 |
Now count what you want to count and get the force field parameters. (I am sorry, the 20 divisions and
natural logarithms you will have to do by pocket calculator).
First check your force field in terms of plausibility. Do the hydrophobic residues have a high
potential for TM helices, and do Arg, Lys, Glu, and ASP indeed score very low as you would expect? If not
dicuss with the assistant what went wrong and fix it.
We did not make any software to score things with your force field, but you can see what happens
if you write the potentials next to the amino acids in this short sequence:
N G T E G P N F Y V P F S N K T G V V R S P F E A P Q Y Y L A
E P W Q F S M L A A Y M F L L I M L G F P I N F L T L Y V T V
Q H K K L R T P L N Y
|
And if all goes well, you will find one TM helix...
The final one.
Question 49:
How would you write software to predict if a residue is an active site residue or not?
Don't forget that active sites are always at the surface of the protein.
Think of the data-set you need.
Think of the null-model.
Think of the real model, and how to convert counting into scoring.
Think about testing the method.
Answer
