
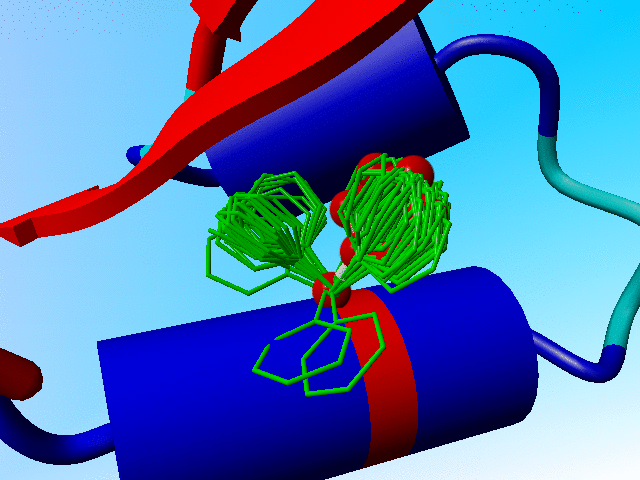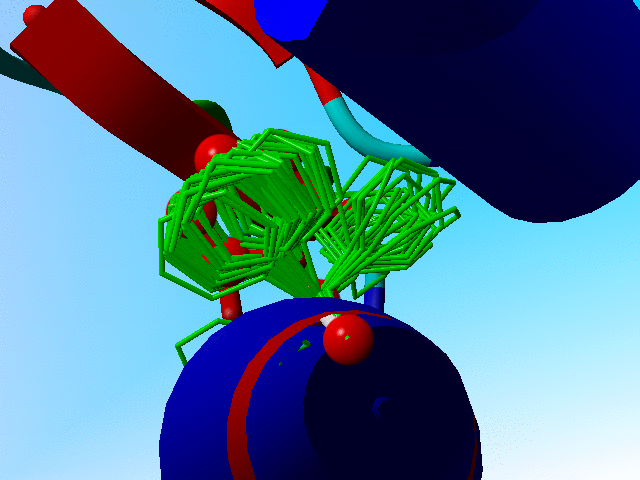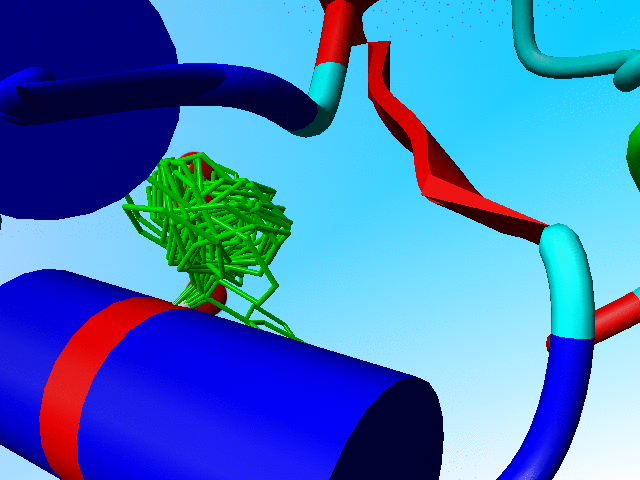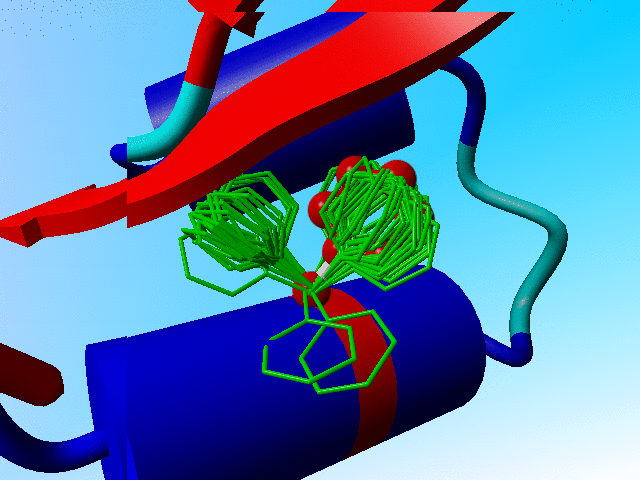
The prefered positions for Phe-13 in crambin.
Proteins aren't rigid bricks. They show a wide variety of modes of motion. One variant is the so called rotamer. Amino acid side chains have prefered conformations, called rotamers. In the next figure I have superposed all low-energy situations for Phenylalanine at position 13 in crambin (energy calculated in the absence of everything but the local backbone of the residues 11 till 13). The two clouds cluster around the two prefered rotamers for Phenylalanine at position 13 in crambin.
|
|
The prefered positions for Phe-13 in crambin. |
In the following blow-up I have added the side chain of phenylalanine as it is actually observed in crambin. You see that it sits "in one of the two prefered rotamers".
|
The observed phenylalanine and the prefered positions for Phe-13 in crambin. |
In the early days of crystallography crystals often weren't very good, X-ray beams were weak, and software wasn't very advanced yet. Therefore, most old PDB files are proteins solved at low resolution, and at low resolution you don't have enough data to start thinking about the possibility that residues can occur in the crystal in two rotamers, or even flip between two or more alternate positions (different rotamers). Consequently, the software was originally not set-up for dealing with alternate conformations, and over the years, needed to be patched for that purpose. It is very obvious to us that this has not gone very well in many, many cases, as is illustrated by the thousands and thousands of problems in dealing with alternate atoms.
In most cases alternate atom problems are purely administrative in nature and can be solved by hard work or by artificially intelligent software. But there is also a fundamental problem. Suppose we have observed two rotamers for a serine, and both occur about 50% of the time (with time I mean either 50% of all molecules in the crystal, or 50% of the time in a dynamic equilibrium, or a combination of both) and these rotamers are called alternate structures A and B. And lets now assume that in both cases another water gets "stuck" to the protein, then we will observe two waters with occupancy 0.5. If at least one of the waters overlaps with one of the serine side chain alternates, we can determine which of the waters should be called A and which should be called B. But if we don't have any such overlap, we need to determine the alternate atom flags for the two water positions using other means than the experimental data.
In the hypothetical example illustrated below, a serine is shown with two alternate positions for its Oγ. There are a lilla and a purple water close to each of the two Oγs. Obviously, these waters are only present together with "the other" Oγ.

|
Serine with two alternates and two waters that bind uniquely to each of the two alternate serine Oγ situations. |
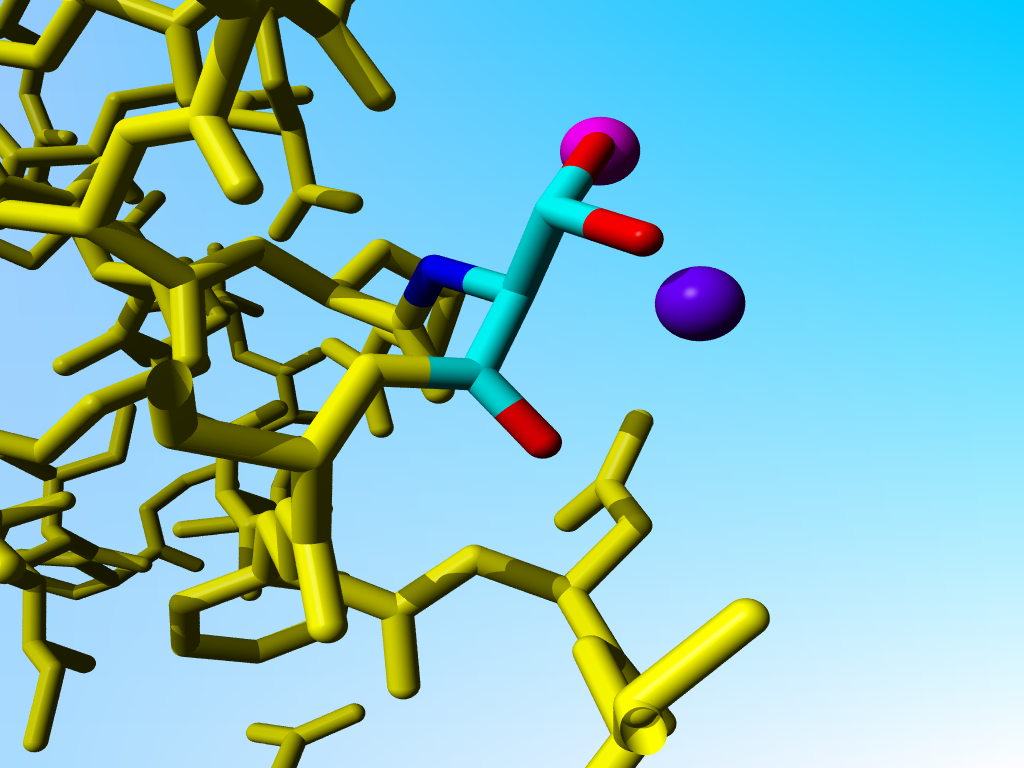
|
Blow-up of the situation around the Serine Oγs in the previous figure. |
EU name: ALTER1
(Date: Aug 24 2016 ALTER1 )
There are many examples of alternate sugar conformations that are not recorded as such, but as fully independent molecules.
REMARK 1 AUTH N.K.VYAS,M.N.VYAS,F.A.QUIOCHO 5ABP 14 REMARK 1 TITL COMPARISON OF THE PERIPLASMIC RECEPTORS FOR 5ABP 15 REMARK 1 TITL 2 L-ARABINOSE-, D-GLUCOSE/D-GALACTOSE-, AND 5ABP 16 REMARK 1 TITL 3 D-RIBOSE. STRUCTURAL AND FUNCTIONAL SIMILARITY 5ABP 17 REMARK 1 REF J.BIOL.CHEM. V. 266 5226 1991 5ABP 18 .... REMARK 1 AUTH F.A.QUIOCHO,N.K.VYAS 5ABP 21 REMARK 1 TITL NOVEL STEREOSPECIFICITY OF THE L-ARABINOSE-BINDING 5ABP 22 REMARK 1 TITL 2 PROTEIN 5ABP 23 REMARK 1 REF NATURE V. 310 381 1984 5ABP 24 REMARK 1 REFN ASTM NATUAS UK ISSN 0028-0836 006 5ABP 25 |
The PDB file 5ABP is just an example. The two sugars are given as independent entities, but both with occupancy 0.5 for all atoms.
Supplemental material
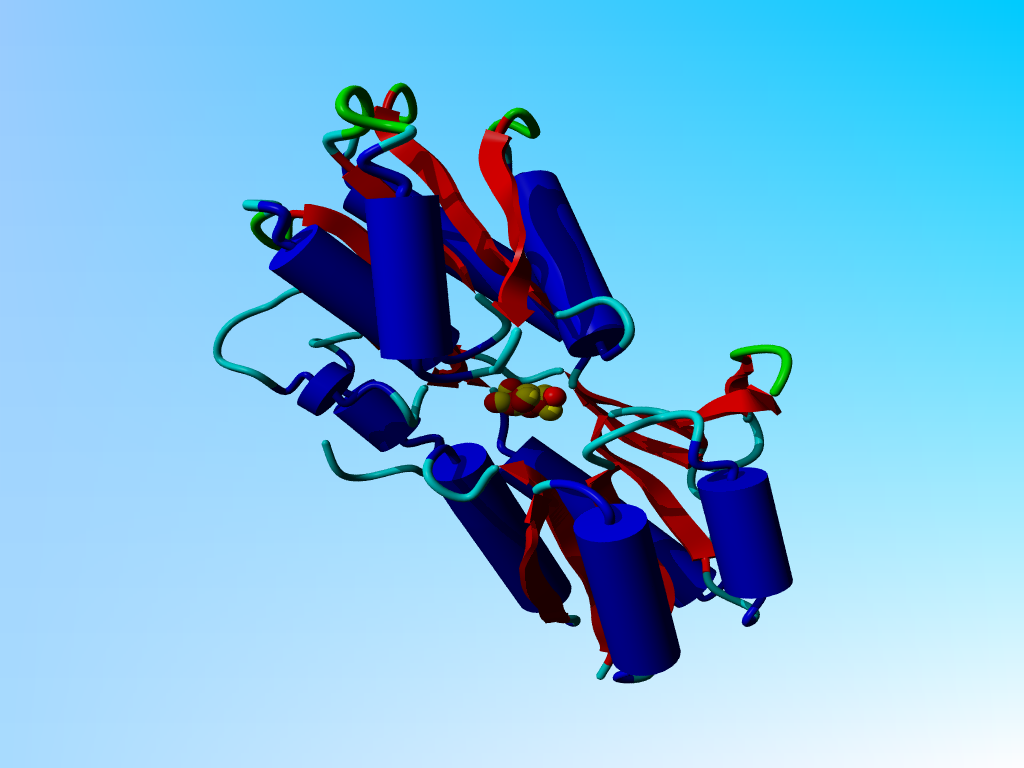
|
5abp with two sugar molecules, one in red and one in yellow. |
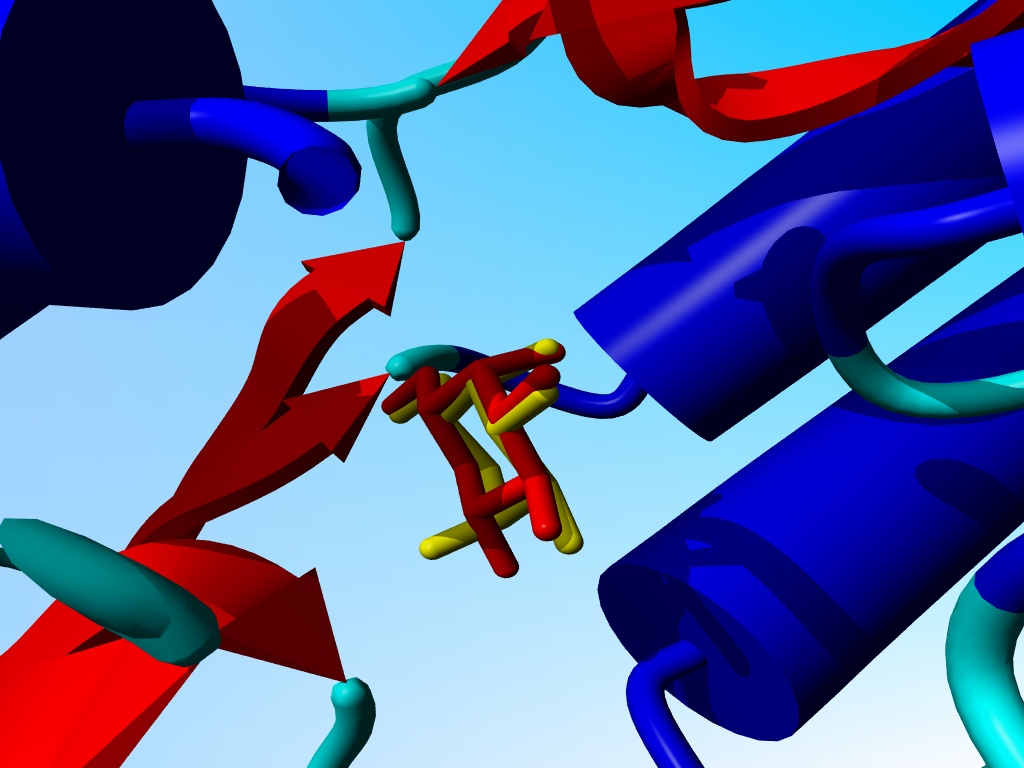
|
Same situation. Click on the picture for a blow-up of the sugars in a stick representation. |
The REMARK cards in the PDB file 5abp suggest that there was no X-ray density to justify the presence of 2 forms of the sugar.
REMARK 5 SINCE IT HAS BEEN SHOWN THAT ABP CAN BIND EITHER ALPHA OR 5ABP 42 REMARK 5 BETA ANOMERS OF D-GALACTOSE WITH ALMOST EQUAL AFFINITY, 5ABP 43 REMARK 5 BOTH ARE PROVIDED IN THE SAME SITE. 5ABP 44 |
But final judgement on the reality of this situation will have to wait for re-refinement of the coordinates. The administrative encoding of the two sugars, however, will confuse many molecular visualisation, calculation, docking, etc. software packages.
JRNL AUTH V.KOENIG,L.VERTESY,T.R.SCHNEIDER JRNL TITL CRYSTAL STRUCTURE OF THE ALPHA-AMYLASE INHIBITOR JRNL TITL 2 TENDAMISTAT AT 0.93 A JRNL REF ACTA CRYSTALLOGR.,SECT.D V. 59 1737 2003 |
This file holds a series of GLA and GLB's (α-D-galactose and β-D-galactose). Two of these (GLA 901 and GLB 902; both in the B chain) overlap nearly completely. The same is true for the C and D chain bound galactoses, but the A chain binds (via its calcium) only to a GLB.
Supplemental material
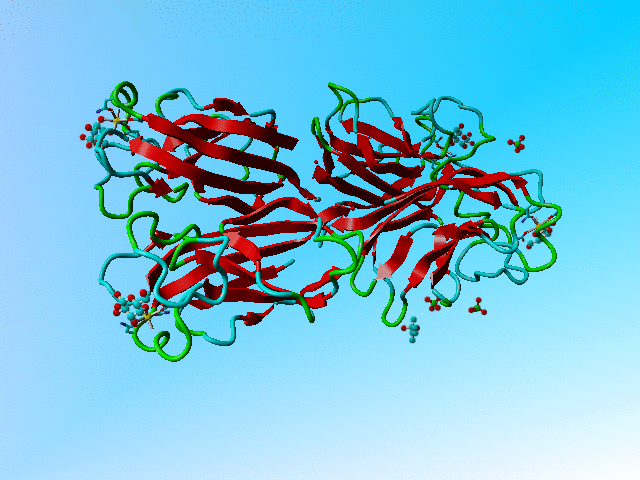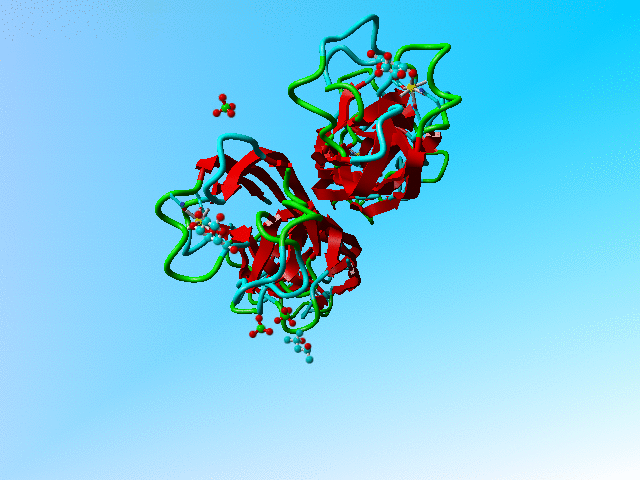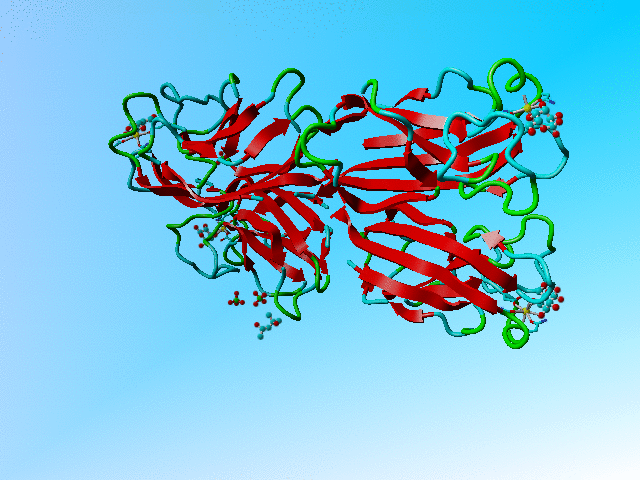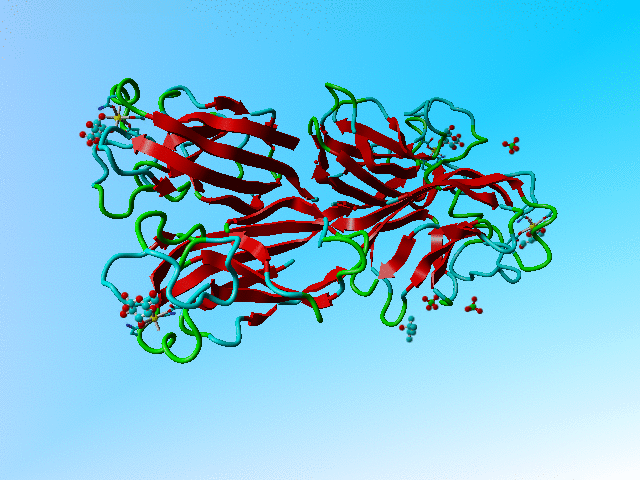
|
The structure of 1oko. Three of the sugars are overlapping GLA and GLB while one is GLB only. |
Supplemental material In all three cases of overlapping sugars with alternate chiralities did the depositors choose two different residues that were placed on top of each other, rather than using one molecule with alternate atoms. Their reasoning might have been that the α and β forms are nor alternates from each other because they have different names, but it would be much easier for all software that works with PDB files to have them as alternates of each other.
JRNL AUTH A.SCHMIDT,C.JELSCH,P.OSTERGAARD,W.RYPNIEWSKI, JRNL AUTH 2 V.S.LAMZIN JRNL TITL TRYPSIN REVISITED: CRYSTALLOGRAPHY AT (SUB) ATOMIC JRNL TITL 2 RESOLUTION AND QUANTUM CHEMISTRY REVEALING DETAILS JRNL TITL 3 OF CATALYSIS. JRNL REF J.BIOL.CHEM. V. 278 43357 2003 |
The molecule 1pq7 contains a bound, free arginine with residue number 703. This arginine has been refined with a rather funny protocoll for as far as the occupancies are concerned. I coloured a few of the most abberant occupancies red in the table below. I also removed the ANISOU records for easier reading. The complete residue is given in the supplemental material listed under the table.
HETATM 3211 N AARG 703 7.213 1.571 -6.245 0.41 19.70 N HETATM 3212 N BARG 703 7.914 0.885 -6.572 0.50 17.49 N HETATM 3213 CA AARG 703 6.341 2.758 -6.523 0.00 26.55 C HETATM 3214 CA BARG 703 7.643 2.395 -6.653 1.08 30.78 C HETATM 3215 C AARG 703 6.769 3.885 -5.588 0.03 29.54 C HETATM 3216 C BARG 703 8.233 3.437 -5.692 0.35 25.04 C HETATM 3217 O AARG 703 6.320 4.392 -4.574 0.55 29.91 O HETATM 3218 O BARG 703 8.410 4.374 -4.819 0.26 7.67 O HETATM 3219 CB AARG 703 4.797 2.546 -6.446 0.69 19.17 C HETATM 3220 CB BARG 703 6.061 2.387 -6.638 0.46 24.15 C HETATM 3221 CG AARG 703 4.078 2.954 -7.714 0.42 19.26 C HETATM 3222 CG BARG 703 5.014 2.302 -7.771 0.25 11.70 C HETATM 3223 CD AARG 703 3.548 1.706 -8.411 0.60 9.71 C HETATM 3224 CD BARG 703 3.874 1.359 -7.501 0.29 10.55 C HETATM 3225 NE AARG 703 3.567 1.539 -9.783 0.30 12.66 N HETATM 3226 NE BARG 703 2.712 1.019 -8.082 0.29 15.91 N HETATM 3227 CZ AARG 703 2.334 1.452 -10.406 0.32 12.44 C HETATM 3228 CZ BARG 703 2.269 1.398 -9.245 0.26 12.19 C HETATM 3229 NH1AARG 703 1.341 1.567 -9.492 0.55 6.14 N HETATM 3230 NH1BARG 703 1.182 1.094 -9.838 0.39 15.81 N HETATM 3231 NH2AARG 703 2.421 1.285 -11.719 0.56 7.40 N HETATM 3232 NH2BARG 703 2.973 2.204 -9.977 0.30 13.94 N |
Supplemental material
Occupancies that don't add up to 1.0 are acceptable to most softwares, but if
an occupancy refines to exactly 0.00 most softwares get into trouble....
EU name: 1A6V
(Date: Aug 24 2016 1A6V )
Occupancies seem to be a complicated process, especially in cases where overlapping ligands are observed. 1A6V is a nice example:
JRNL AUTH T.SIMON,K.HENRICK,M.HIRSHBERG,G.WINTER JRNL TITL X-RAY STRUCTURES OF FV FRAGMENT AND ITS JRNL TITL 2 (4-HYDROXY-3-NITROPHENYL)ACETATE COMPLEX OF MURINE JRNL TITL 3 B1-8 ANTIBODY JRNL REF TO BE PUBLISHED |

|
The ligands NPC-H-430 and NPC-H-431 have different names, but seem to be alternates for each other. Here they are drawn in yellow and red respectively. The two other ligands in the background are two NPCs in two other chains in the asymmetric unit. |
HETATM 5239 C1 NPC H 430 7.065 12.471 75.277 0.50 13.83 HETATM 5240 C2 NPC H 430 5.935 12.505 76.061 0.50 13.71 HETATM 5241 C3 NPC H 430 5.124 11.383 76.140 1.00 14.48 HETATM 5242 N3 NPC H 430 3.988 11.450 76.958 1.00 14.16 HETATM 5243 O3A NPC H 430 3.635 12.555 77.429 1.00 19.79 HETATM 5244 O3B NPC H 430 3.351 10.404 77.200 1.00 14.67 HETATM 5245 C4 NPC H 430 5.457 10.217 75.430 1.00 15.44 HETATM 5246 O4 NPC H 430 4.630 9.065 75.494 1.00 15.86 HETATM 5247 C5 NPC H 430 6.598 10.184 74.663 0.50 14.33 HETATM 5248 C6 NPC H 430 7.415 11.312 74.584 0.50 12.58 HETATM 5249 C7 NPC H 430 7.899 13.647 75.151 0.50 15.05 HETATM 5250 C8 NPC H 430 7.919 14.392 73.868 0.50 14.50 HETATM 5251 O8 NPC H 430 7.726 13.840 72.767 0.50 15.95 HETATM 5252 N9 NPC H 430 8.153 15.659 73.916 0.50 15.56 HETATM 5253 C10 NPC H 430 9.241 16.188 73.203 0.50 15.67 HETATM 5254 C11 NPC H 430 8.866 17.553 72.623 0.50 16.65 HETATM 5255 C12 NPC H 430 8.992 17.525 71.008 0.50 18.67 HETATM 5256 C13 NPC H 430 9.110 18.987 70.441 0.50 20.57 HETATM 5257 C14 NPC H 430 10.559 19.137 69.913 0.50 22.47 HETATM 5258 C15 NPC H 430 11.396 18.038 69.695 0.50 23.82 HETATM 5259 O15 NPC H 430 11.546 17.083 70.484 0.50 25.87 HETATM 5260 O16 NPC H 430 12.068 17.990 68.617 0.50 27.69 HETATM 5261 C1 NPC H 431 7.506 13.112 75.302 0.50 15.56 HETATM 5262 C2 NPC H 431 6.227 13.136 76.024 0.50 15.68 HETATM 5263 C3 NPC H 431 5.320 11.905 76.150 0.50 16.39 HETATM 5264 N3 NPC H 431 4.083 12.016 76.921 0.50 14.56 HETATM 5265 O3A NPC H 431 3.648 13.147 77.345 0.50 18.48 HETATM 5266 O3B NPC H 431 3.449 10.998 77.175 0.50 16.88 HETATM 5267 C4 NPC H 431 5.759 10.735 75.575 0.50 18.19 HETATM 5268 O4 NPC H 431 4.993 9.609 75.724 0.50 17.01 HETATM 5269 C5 NPC H 431 7.106 10.741 74.898 0.50 14.32 HETATM 5270 C6 NPC H 431 7.949 11.907 74.799 0.50 14.22 HETATM 5271 C7 NPC H 431 8.312 14.338 75.054 0.50 17.01 HETATM 5272 C8 NPC H 431 7.835 15.288 74.019 0.50 14.94 HETATM 5273 O8 NPC H 431 7.415 16.393 74.393 0.50 14.67 HETATM 5274 N9 NPC H 431 7.759 14.908 72.778 0.50 16.12 HETATM 5275 C10 NPC H 431 8.044 15.795 71.734 0.50 18.32 HETATM 5276 C11 NPC H 431 9.383 15.406 71.099 0.50 20.54 HETATM 5277 C12 NPC H 431 9.177 15.015 69.536 0.50 20.85 HETATM 5278 C13 NPC H 431 9.586 13.518 69.291 0.50 21.74 HETATM 5279 C14 NPC H 431 8.326 12.673 68.968 0.50 22.57 HETATM 5280 C15 NPC H 431 7.616 13.403 67.778 0.50 23.06 HETATM 5281 O15 NPC H 431 8.176 14.058 67.017 0.50 24.28 HETATM 5282 O16 NPC H 431 6.381 13.091 67.672 0.50 22.48 |
Pay special attention to the red atoms in NPC 430. One would expect the alternates to have occupancy zero and exactly identical coordinates. Unfortunately, the equivalent green atoms are about one or two Ångström away from the red ones, and have occupancy 0.5 which means that they are 1.5 times present....
EU name: ALTER2
(Date: Aug 24 2016 ALTER2 )
Although there are no rules for this, it seems good practice to place alternate copies of the same molecule directly consequetively in the PDB file. For a serine, for example, this goes fine as it is obvious that when you see two Oγs in the denisty that they are both alternate atoms that are bound to the same Cβ. But for water atoms this is a problem as one never knows which water is an alternate for which water. An example of an unelegant solution is found in 1XG0:
JRNL AUTH A.B.DOUST,C.N.J.MARAI,S.J.HARROP,K.E.WILK, JRNL AUTH 2 P.M.G.CURMI,G.D.SCHOLES JRNL TITL DEVELOPING A STRUCTURE-FUNCTION MODEL FOR THE JRNL TITL 2 CRYPTOPHYTE PHYCOERYTHRIN 545 USING ULTRAHIGH JRNL TITL 3 RESOLUTION CRYSTALLOGRAPHY AND ULTRAFAST LASER JRNL TITL 4 SPECTROSCOPY JRNL REF J.MOL.BIOL. V. 344 135 2004 |
In this file I find, for example, the following two waters:
... HETATM 5037 O HOH 953 -15.302 30.693 7.141 0.50 12.57 O ... HETATM 5069 O HOH 985 -14.838 30.632 7.560 0.50 32.72 O |
These two waters are 0.63 Ångström away from each other, but they are 60 lines
away from each other in the PDB file.
EU name: ALTER3
(Date: Aug 24 2016 ALTER3 )
JRNL AUTH C.N.FUHRMANN,B.A.KELCH,N.OTA,D.A.AGARD JRNL TITL THE 0.83A RESOLUTION CRYSTAL STRUCTURE OF JRNL TITL 2 ALPHA-LYTIC PROTEASE REVEALS THE DETAILED JRNL TITL 3 STRUCTURE OF THE ACTIVE SITE AND IDENTIFIES A JRNL TITL 4 SOURCE OF CONFORMATIONAL STRAIN. |
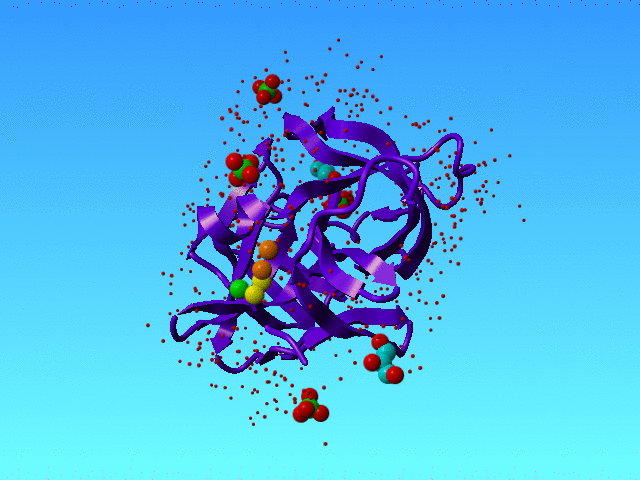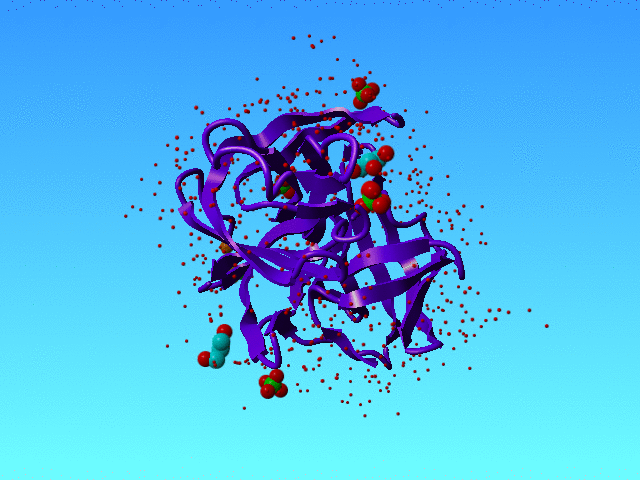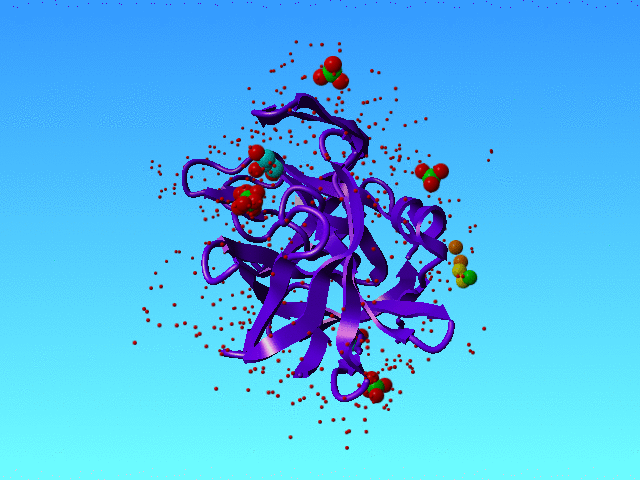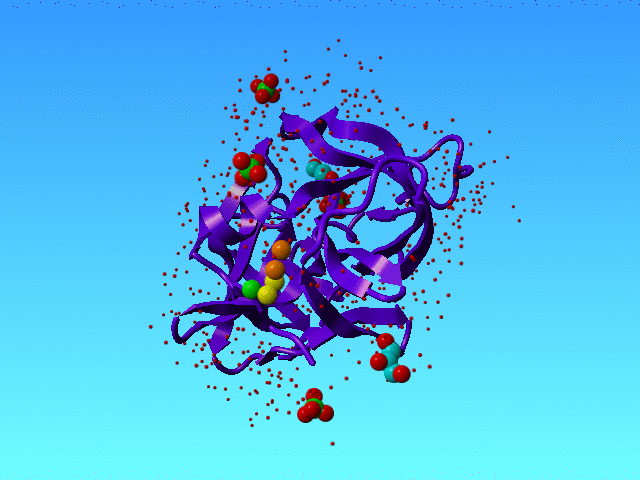
In 1SSX we find many alternate atoms. That is not surprising because this structure has been solved at 0.83 Angstrom resolution, and that is high enough to see very many alternate condformations.
|
1SSX is solved at high resolution, and that allows the authors to see many waters and many alternate conformations, and even alternate water conformations. The green, yellow, and orange spheres are three water atoms that we discuss here. The protein is a purple ribbon. The small red dots are waters that are not discussed in this section. |
However, our software got a bit confused when it ran into the waters that in 1SSX are numbered 615, 616, and 639. Actually, it got confused about many waters, but these three I will explain as an example.
HETATM 3477 O AHOH 615 44.531 24.042 19.491 0.50 11.79 HETATM 3478 O BHOH 615 44.058 25.531 19.822 0.50 15.98 HETATM 3479 O AHOH 616 43.993 26.552 20.613 0.50 18.33 O HETATM 3480 O BHOH 616 43.014 28.390 21.383 0.20 19.39 HETATM 3509 O HOH 639 44.007 23.243 21.026 0.50 25.10 |
The colours in the table correspond with the colours in the figure above.
Striktly formally speaking this situation is correct (I think) because all space is filled with maximally 1 water, so there are no clashes. But the green water can only be present when the lower yellow is not there, and the lower orange water can only be there when the higher yellow one is not there. So in a way, we have some soliton wave of waters here: If the orange one moves to the bottom situation then the yellow must do that too, and the green one is kicked into the bulk; and when the green one kicks back in, the yellow one moves to the top position, and kicks the orange partly out to the bulk and partly to the top orange position. So, in a way, these waters are all alternates for each other.
I hope you forgive us that we have no idea yet how to deal with such cases. Working out the hydrogen bonding network
for this molecule simply is impossible with the computer power available today (and tomorrow).
EU name: ALTER5
(Date: Aug 24 2016 ALTER5 )
JRNL AUTH I.S.RIDDER,H.J.ROZEBOOM,B.W.DIJKSTRA JRNL TITL HALOALKANE DEHALOGENASE FROM XANTHOBACTER JRNL TITL 2 AUTOTROPHICUS GJ10 REFINED AT 1.15 A RESOLUTION. JRNL REF ACTA CRYSTALLOGR.,SECT.D V. 55 1273 1999 |
The file 1B6G has been solved at a beautiful 1.15 Ångström. At such high resolution one can, obviously, detect many alternates. But the alternate flag for Pb in the lead-bound cysteine 150 in this file is a bit surprising. As the reflections have not been deposited for this file, we cannot do much further than being confused about this Pb.
HETATM 1290 N CSB A 150 36.630 29.004 32.204 1.00 10.18 N HETATM 1291 CA CSB A 150 37.436 29.544 33.307 1.00 10.02 C HETATM 1292 CB CSB A 150 37.680 31.055 33.172 1.00 10.63 C HETATM 1293 SG CSB A 150 36.152 32.054 33.239 1.00 13.36 S HETATM 1294 PB BCSB A 150 34.802 32.151 34.168 0.14 10.61 PB HETATM 1295 C CSB A 150 36.791 29.102 34.618 1.00 10.20 C HETATM 1296 O CSB A 150 36.035 28.130 34.639 1.00 11.76 O |
EU name: 3CI3
(Date: Aug 24 2016 3CI3 )
JRNL AUTH M.ST MAURICE,P.MERA,K.PARK,T.C.BRUNOLD, JRNL AUTH 2 J.C.ESCALANTE-SEMERENA,I.RAYMENT JRNL TITL STRUCTURAL CHARACTERIZATION OF A HUMAN-TYPE JRNL TITL 2 CORRINOID ADENOSYLTRANSFERASE CONFIRMS THAT JRNL TITL 3 COENZYME B12 IS SYNTHESIZED THROUGH A JRNL TITL 4 FOUR-COORDINATE INTERMEDIATE. JRNL REF BIOCHEMISTRY 2008 |
Sometimes it is hard to understand where occupancies come from. The
3ci3
PDB file 3CI3
has been solved at 1.11 Ångström. Looking at
ATOM 670 N ASER A 83 -5.247 -27.565 -12.945 0.50 23.99 N ATOM 671 N BSER A 83 -5.265 -27.477 -12.962 0.50 20.94 N ATOM 672 CA ASER A 83 -6.675 -27.054 -13.202 0.00 19.92 C ATOM 673 CA BSER A 83 -6.620 -26.991 -13.212 0.50 17.74 C ATOM 674 C ASER A 83 -6.878 -25.735 -12.331 0.50 16.97 C ATOM 675 C BSER A 83 -7.066 -26.018 -12.139 0.50 17.74 C ATOM 676 O ASER A 83 -6.210 -25.621 -11.298 0.50 17.49 O ATOM 677 O BSER A 83 -6.498 -25.998 -11.045 0.50 17.42 O ATOM 678 CB ASER A 83 -7.639 -28.237 -13.081 0.00 19.97 C ATOM 679 CB BSER A 83 -7.584 -28.152 -13.254 0.50 18.52 C ATOM 680 OG ASER A 83 -7.362 -29.224 -14.058 0.00 20.41 O ATOM 681 OG BSER A 83 -7.552 -28.728 -11.996 0.50 20.91 O |
This is funny. First, although this is not a rule, it would have been wiser to use the alternate B for the version with the missing atoms because many visualizers use only version A and throw 'higher' versions away. And second, there is density for version B. Perhaps not enough for an occupancy of 0.5, but the B-alternate atoms are in highly acceptable density:
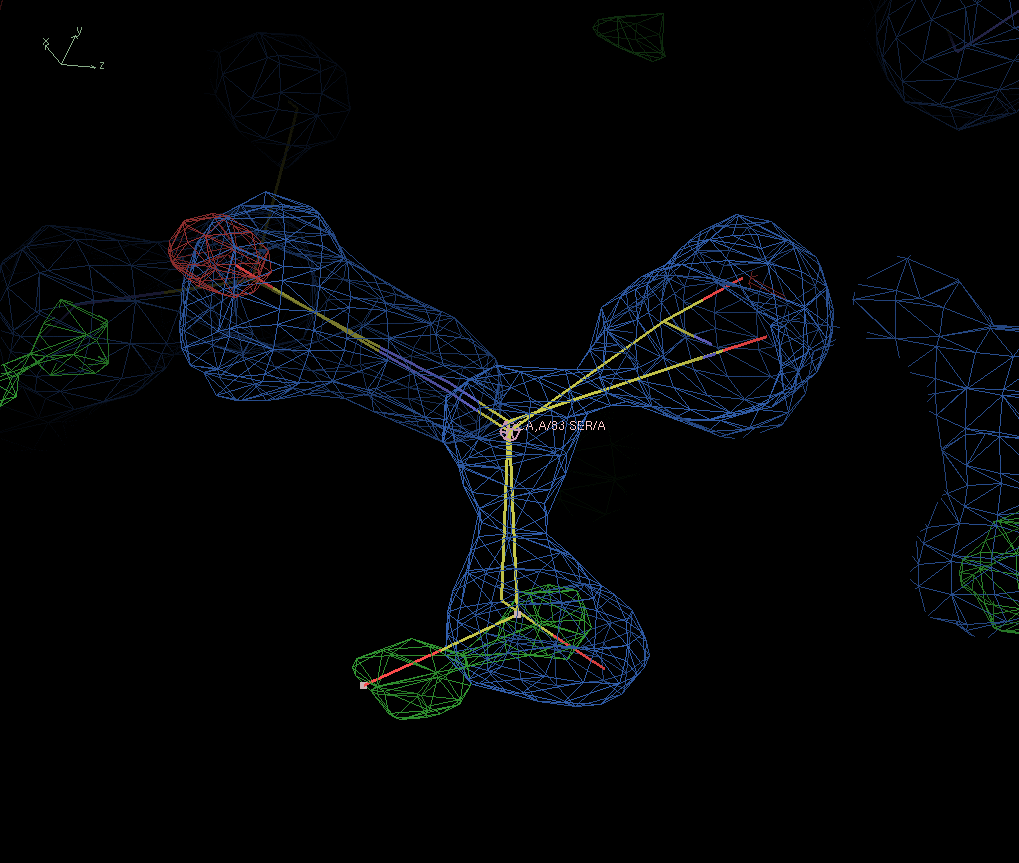
|
The two alternates of ser-83 in 3IC3. The green density is density in which not enough atoms were found. Which makes sense because the Oγ that sits in this density has occupancy zero. |
EU name: ALTER4
(Date: Aug 24 2016 ALTER4 )
Talking of alternate atoms, by the way, I was struck by the lack of an ontology for those. So, on April 1 2009 I look at all atoms in the PDB for which one atom and one alternate atom have been given. I divided these pairs in three classes:
In the three lists below, the alternate atom indicator pair is given (- stands for a blank) in the order they were found in the PDB. Things one could call crazy like one atom having an alternate atom indicator and the other one not, or the first one listed having an alternate atom indicator that comes in the alphabet after the one used for the second alternate listed, etc., are rare. But there is fundamentally no (detectable) ontology for the alternate atom indicators that can be used.
First occupancy larger -A 1 AB 168344 AC 216 BC 201 AD 14 BD 10 CD 168 CE 1 DE 11 DF 45 GH 3 AI 10 IJ 7 KL 1 AO 20 NO 3 OP 8 GR 54 LR 1 ST 4 UV 4 12 803 |
Occupancies equal BA 2 2A 9 AB 415885 -B 2 AC 276 BC 460 BD 112 CD 444 DE 100 EF 72 EG 20 FH 20 AN 1 CO 378 QR 7 BS 1 PS 21 IY 51 YZ 120 12 1186 -- 2 |
Second occupancy larger AB 45006 AC 274 BC 220 AD 2 BD 9 CD 29 DE 7 EF 9 GH 8 CN 6 MN 4 CO 126 LU 104 12 323 |
These tables contain only atom pairs that were detected on consequetive lines in
PDB files.
EU name: 2E86
(Date: Aug 24 2016 2E86 )
Sometimes it is hard to imagine why things happen they happen. Take, for example, Arg-187 in 2e86 (as I did on 8 Mar 2012).
JRNL AUTH E.I.TOCHEVA,L.D.ELTIS,M.E.P.MURPHY JRNL TITL CONSERVED ACTIVE SITE RESIDUES LIMIT INHIBITION OF A JRNL TITL 2 COPPER-CONTAINING NITRITE REDUCTASE BY SMALL MOLECULES. JRNL REF BIOCHEMISTRY V. 47 4452 2008 |
This residue has very weird coordinates:
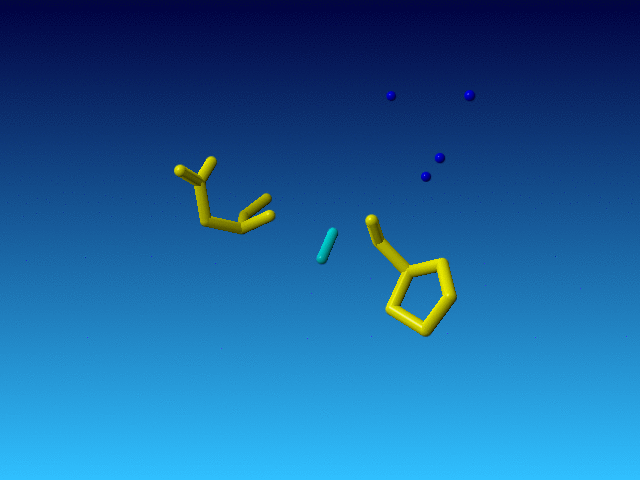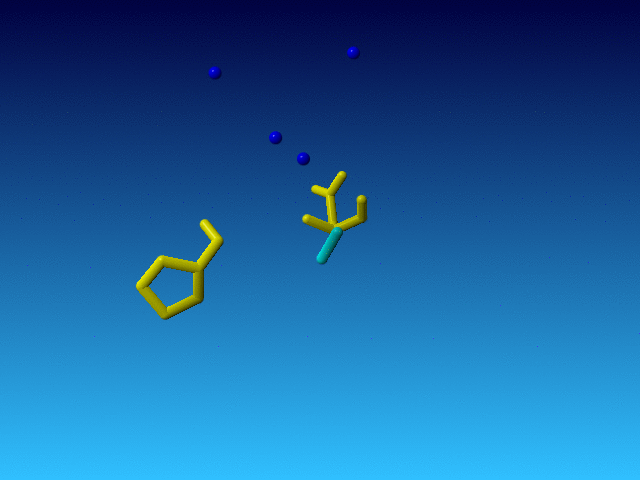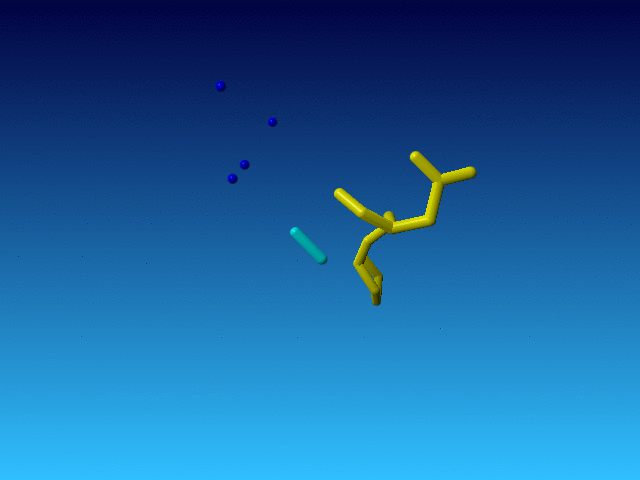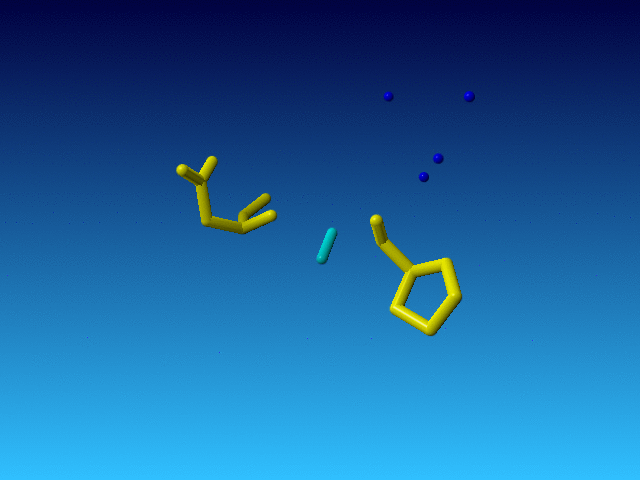
|
The Pro before this Arg is normal, and so is the Asp behind it in the sequence. But the Arg is very incomplete. And what is even weirder, the Arg is the only residue in the whole file that has alternate atoms...: |
ATOM 6544 N PRO C 186 43.420 47.831 39.942 1.00 12.94 N ATOM 6545 CA PRO C 186 44.687 47.486 40.601 1.00 13.12 C ATOM 6546 C PRO C 186 45.405 48.682 41.232 1.00 13.30 C ATOM 6547 O PRO C 186 45.293 49.803 40.731 1.00 13.50 O ATOM 6548 CB PRO C 186 45.529 46.917 39.456 1.00 13.38 C ATOM 6549 CG PRO C 186 44.528 46.419 38.468 1.00 13.20 C ATOM 6550 CD PRO C 186 43.412 47.412 38.528 1.00 12.93 C ATOM 6551 CA ARG C 187 47.825 47.392 45.472 0.50 39.66 C ATOM 6552 CB ARG C 187 48.353 47.100 44.053 0.50 39.65 C ATOM 6553 NH1AARG C 187 44.012 50.671 50.582 0.50 32.31 N ATOM 6554 NH1BARG C 187 40.783 50.820 46.892 0.50 10.32 N ATOM 6555 NH2AARG C 187 43.486 47.581 47.739 0.50 20.93 N ATOM 6556 NH2BARG C 187 42.659 48.382 47.403 0.50 25.47 N ATOM 6557 N ASP C 188 49.338 50.085 43.113 1.00 29.42 N ATOM 6558 CA ASP C 188 50.723 49.757 43.448 1.00 29.58 C ATOM 6559 C ASP C 188 50.936 49.607 44.960 1.00 29.71 C ATOM 6560 O ASP C 188 49.987 49.714 45.741 1.00 29.63 O ATOM 6561 CB ASP C 188 51.690 50.794 42.851 1.00 29.57 C ATOM 6562 CG ASP C 188 51.400 52.219 43.314 1.00 29.49 C ATOM 6563 OD1 ASP C 188 51.045 52.423 44.496 1.00 28.85 O ATOM 6564 OD2 ASP C 188 51.546 53.145 42.487 1.00 30.11 O |