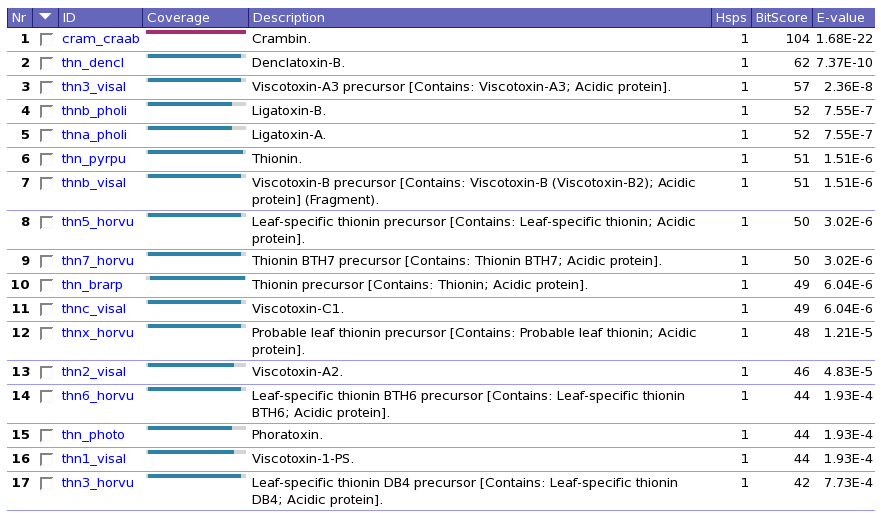
Figure 35. Example BLAST output.
|
After completing the "BLAST" part of the Tools section you: |
Homology and similarity are two basic elements that will pop up many times throughout this course. If we want to find similar sequences, we need to compare one sequence with all other sequences in a database. The best way of performing such a database search would, of course, be a pairwise sequence alignment of the query sequence with each of the sequences in the database. In practice this is costly because either you have to wait a loooooong time, or you have to buy a very big computer or a dedicated sequence alignment computer. Several people have therefore designed methods that are very fast in selecting potential candidate sequences so that the list of sequences that needs to be analysed using a pairwise sequence alignment is relatively short. Of course, the filter method should neither be too strict (we don't want to lose good candidates in the filtering) nor should it be to lax (we don't want to align too many bad candidates).
Two of the best known programs for this database-searching are FASTA and BLAST. In this course we shall use BLAST. BLAST is a program that compares one sequence (the so-called query sequence ) with all sequences in a database. BLAST has some built-in tricks that speed it up. These tricks will cause it to occasionally miss a database file, but it never misses a database file that has high sequence similarity . So BLAST is the ideal compromise between speed and quality, and consequently, BLAST is these days the most used sequence database search program.
In ten years, we will undoubtedly do database searches with a newer, better program, but that program's user interface will have many characteristics of the BLAST interface:
EU name: BLASTO
(From: ../EUDIR )
(Date: Jan 27 17:59 ../EUDI)
Lets start playing with the MRS version of BLAST. You activate this BLAST by clicking on the word Blast in the list of MRS facilities (in the menu near the top of the page).
Do a BLAST search with the following sequence:
TTCCPSIVARSNFNVCRLPGTPEAICATYTGCIIIPGATCPGDYAN |
Hint: You can just cut-and-paste the sequence from the webpage into the MRS-BLAST server
window, and make it FASTA format by adding the > sign and a title, or you can ask
MRS-BLAST to do that for you simply by clicking 'Run Blast'. WARNING: This automatic
conversion from just-a-sequence to a FASTA-format-sequence potentially can
reset a series of Blast parameters, so, first fix the sequence format and thereafter think
about all Blast-parameters).
If everything goes right, you will get something that (very roughly) looks like the
pictures below.
|
|
Figure 35. Example BLAST output. |
Question 17: Annotate the BLAST output you obtained; and whith annotate we mean that you should describe at laest the content of each column.
AnswerIf you now click in that BLAST output on any coloured bar in the column "coverage", and subsequently on the double coloured bar that pops up and that represents the coverage of the sequence pair, you get to see approximately:
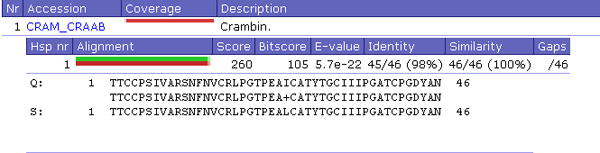
|
Figure 36. Example BLAST output for one hit. |
Question 18: Annotate this picture too.
Answer
Question 19:
Run BLAST on the sequence of the SwissProt file CRAM_CRAAB against the SwissProt database.
(obviously, the first hit will be CRAM_CRAAB...). Write down the Bitscore and the E-value.
Run BLAST with on the sequence of the SwissProt file CRAM_CRAAB against the Uniprot KB database.
Write down the Bitscore and the E-value.
Do you understand the four numbers you just wrote down? Hint:
How many sequences do the SwissProt database and the UniProt database contain?
Please explain the relation between the E-values and the size of the database.
EU name: BLAST3
(From: ../EUDIR )
(Date: Jan 27 17:59 ../EUDI)
BLAST is the central vehicle in our quest for (transfer of) information. In the next three BLAST exercises you will each time be asked to find a residue number. At the end you will do some math with those numbers to end up with The answer.
So you are not supposed to find exact sequences, bur carry-over information from homologous sequences to each of the three query sequences.
Which is the bridged cysteine (that is annotated as disufid) with the lowest number in the boxed sequence? So find the cysteine that has a disulfide bridge with another cysteine, and that has the lowest number in its own sequence.
SAANILGKEAKCTDQVNGCPRIFNPVCGTEGVTYSNECLICMENKREQTPVLIERSGPC |
How many residues bind the Zinc in:
FLPYNALASTEHVTWNQQFQTPQFISGDLLKVNGTSPEELVYQYVEKNENKFKFHENAK DTLQLKEKKNDNLGFTFMRFQQTYKGIPVFGAVVTSHVKDGTLTALSGTLIPNLDTKGS LKSGKKLSEKQARDIAEKDLVANVTKEVPEYEQGKDTEFVVYVNGDEASLAYVVNLNFL TPEPGNWLYIIDAVDGKILNKFNQLDAAKPGDVKSITGTSTVGVGRGVLGDQKNINTTY STYYYLQDNTRGNGIFTYDAKYRTTLPGSLWADADNQFFASYDAPAVDAHYYAGVTYDY YKNVHNRLSYDGNNAAIRSSVHYSQGYNNAFWNGSQMVYGDGDGQTFIPLSGGIDVVAH ELTHAVTDYTAGLIYQNESGAINEAISDIFGTLVEFYANKNPDWEIGEDVYTPGISGDS LRSMSDPAKYGDPDHYSKRYTGTQDNGGVHINSGIINKAAYLISQGGTHYGVSVV |
A friend made a mistake cloning the sequence in the next box. His cloned sequence is too short. Can you tell him how many residues are missing?
MAHAWGPQRLAGGQPQANFEESTQGSIFTYTNSNSTRDPFEGPNYHIAPRWVYHLTSAWM VFVVIASVFTNGLVLAATMRFKKLRHPLNWILVNLAIADLAETIIASTISVVNQMYGYFV LGHPLCVVEGYTVSLCGITGLWSLAIISWERWMVVCKPFGNVRFDAKLAITGIAFSWIWA AVWTAPPIFGWSRYWPHGLKTSCGPDVFSGSSYPGVQSYMIVLMITCCFIPLSVIILCYL QVWLAIRAVAKQQKESESTQKAEKEVTRMVMVMIFAYCLCWGPYTFFACFAAAHPGYAFH PLVAALPAYFAKSATIYNPIIYVFMNRQFRNCILQLF |
Question 20: And now the math. Brace yourself, get the calculators out. What do you get if you add up the three requested numbers?
Answer
EU name: TRNSOS
(From: ../EUDIR )
(Date: Jan 27 17:59 ../EUDI)
In the examples above, we looked for direct, hard data. Sometimes data isn't that hard...
Question 21: In the MRS section you studied the crambin protein. With the help of the BLAST output of crambin (you should by now know how to get hold of this sequence...), you should be able to come up with new/further properties of crambin.
Which is the likely function of crambin?
Answer