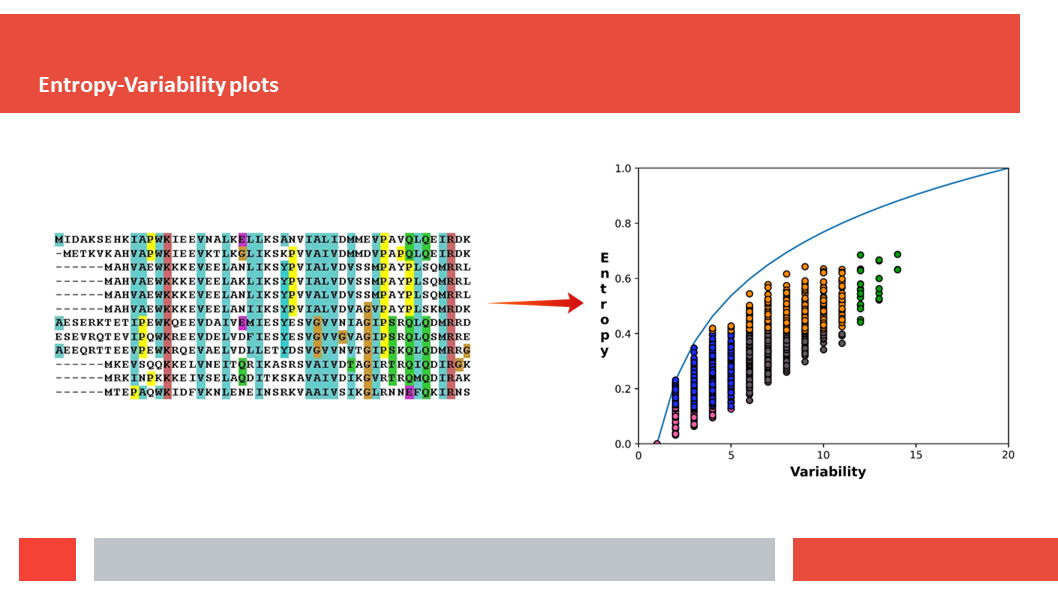
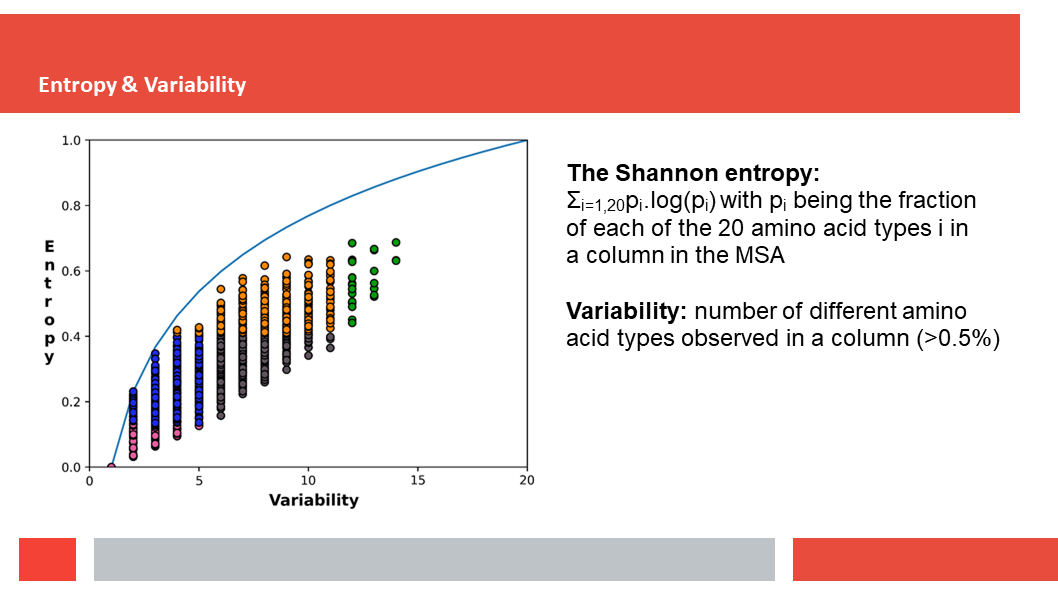
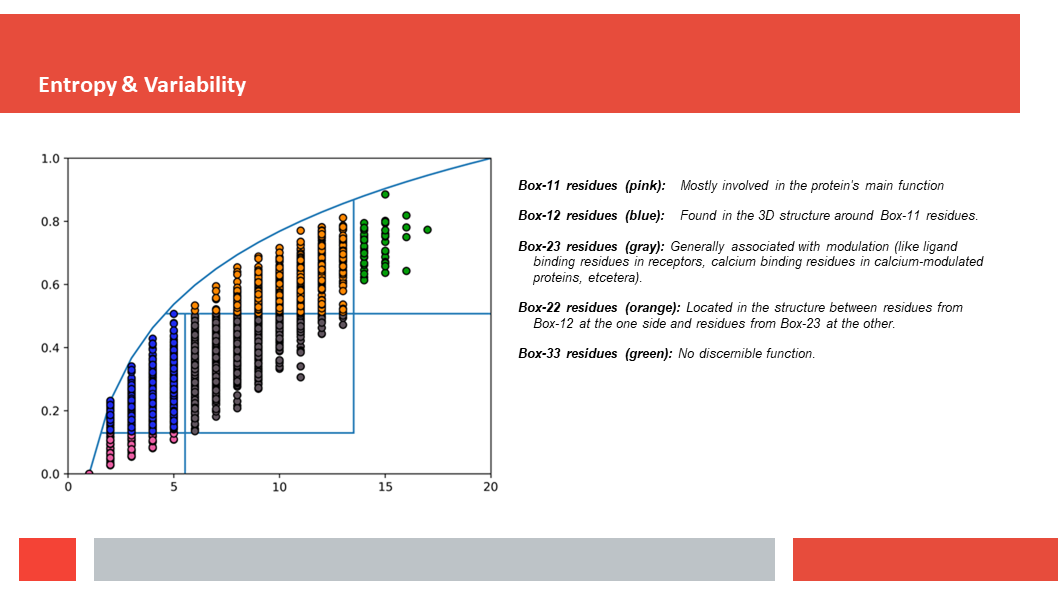
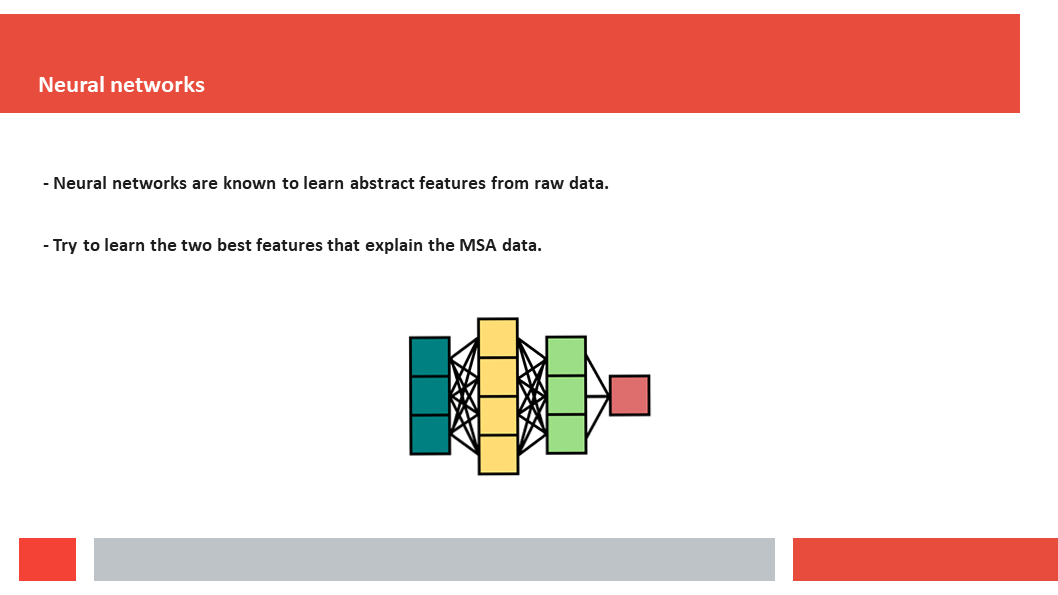
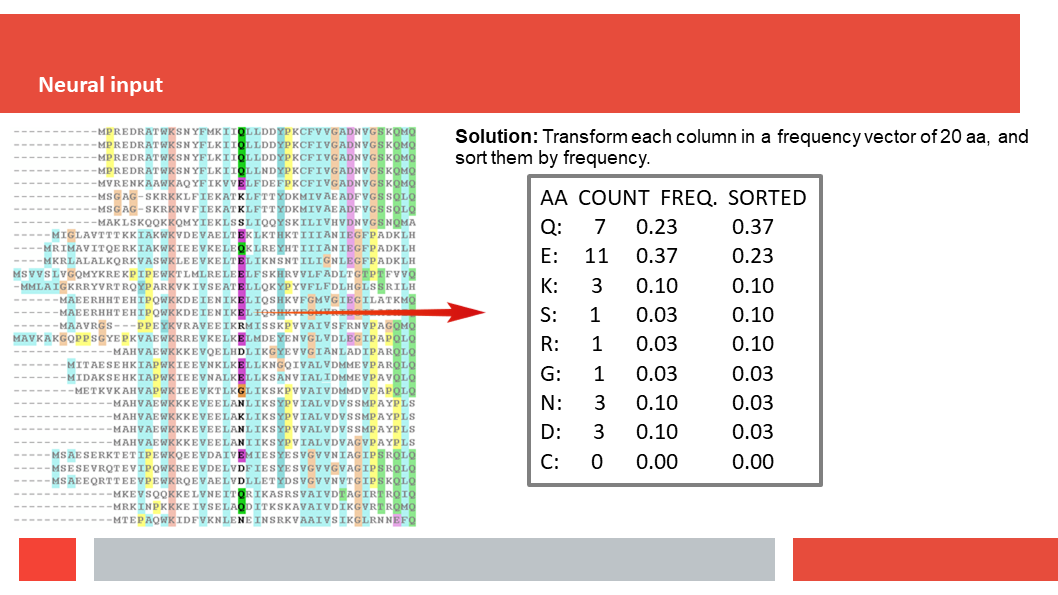
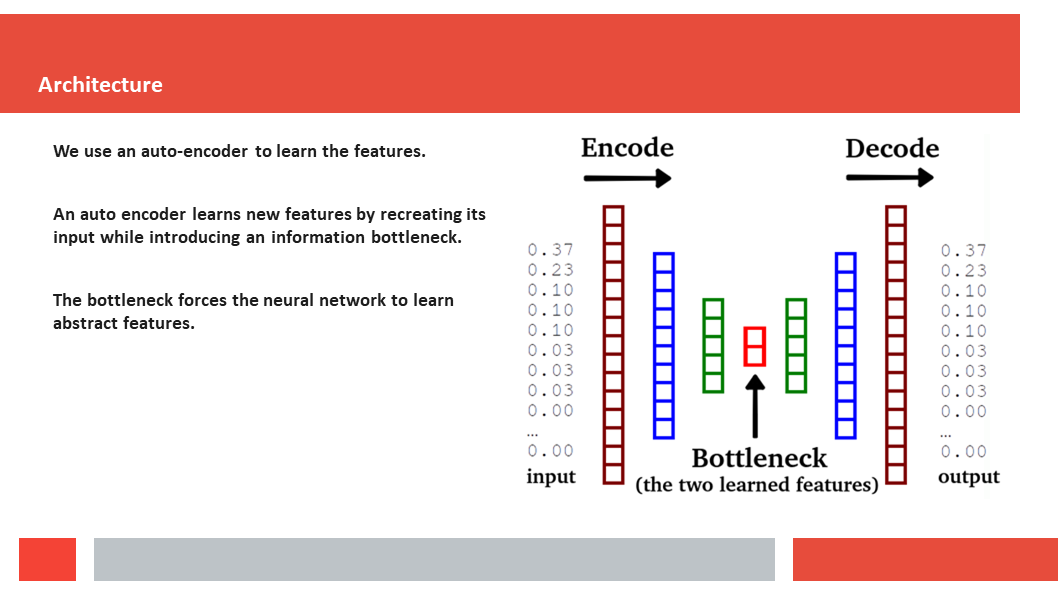
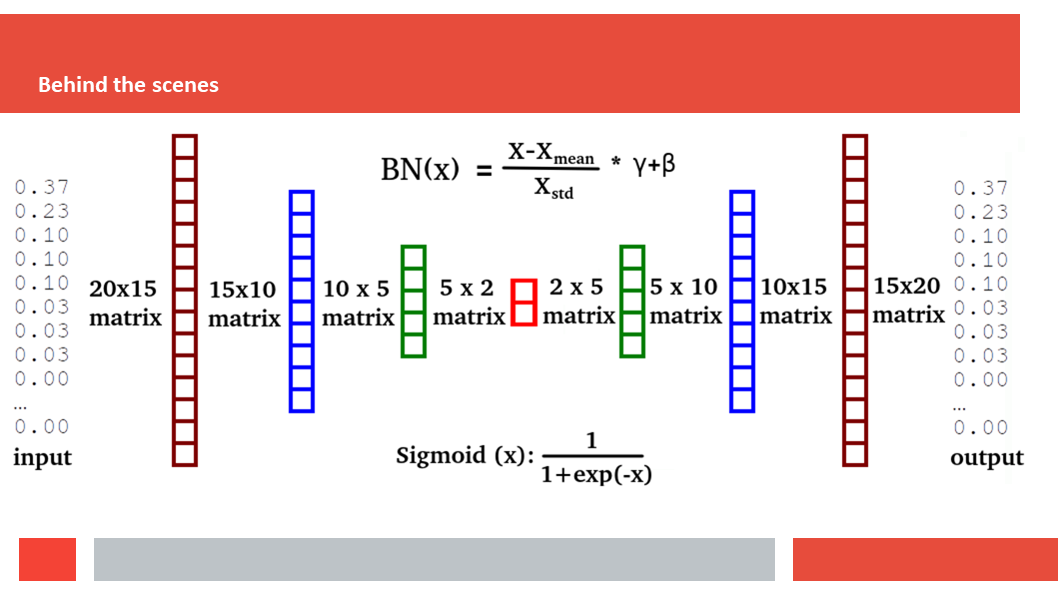
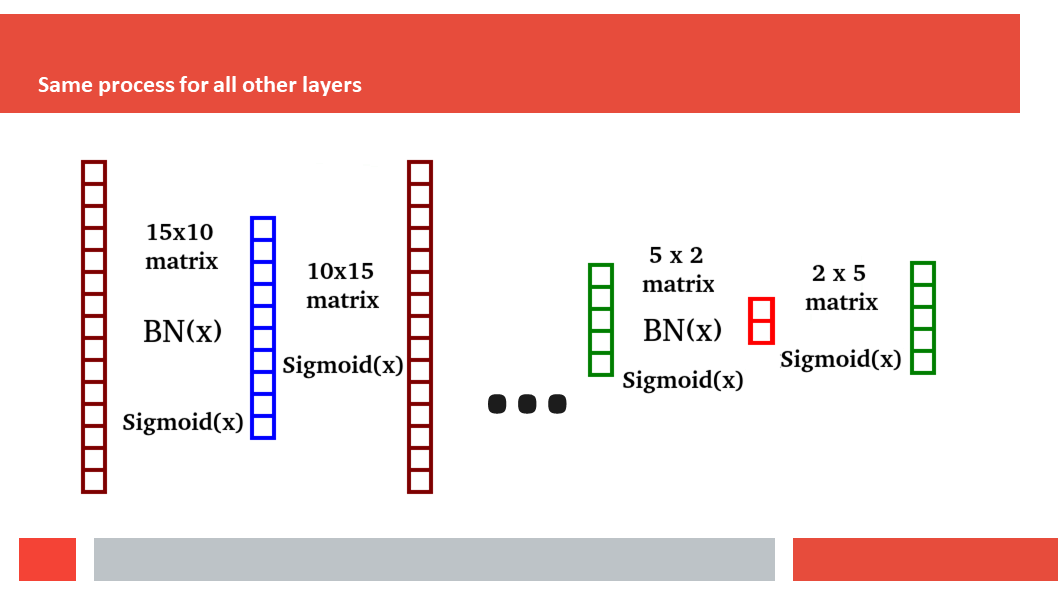
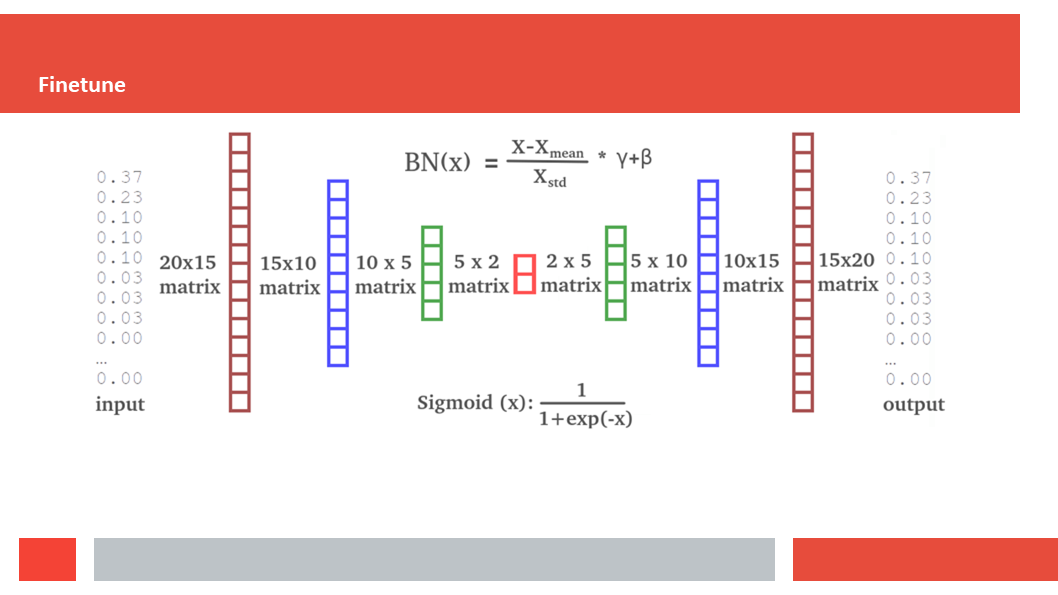
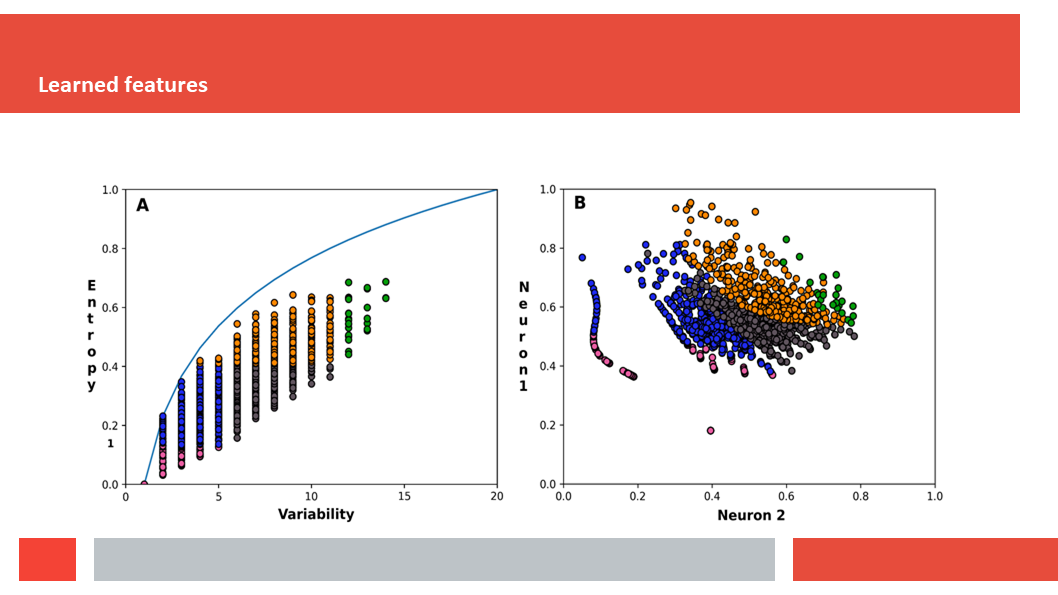
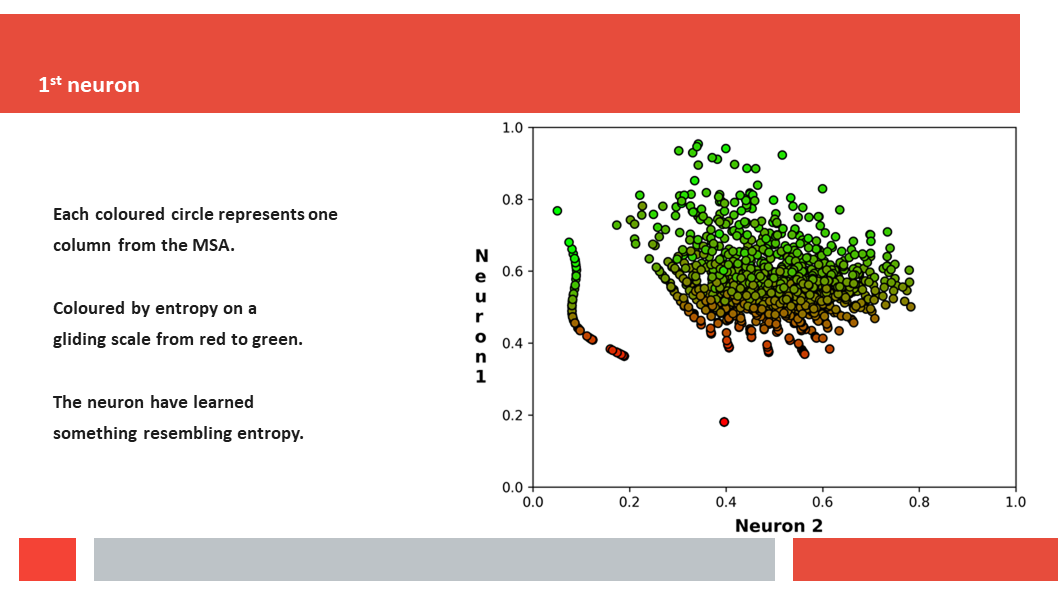
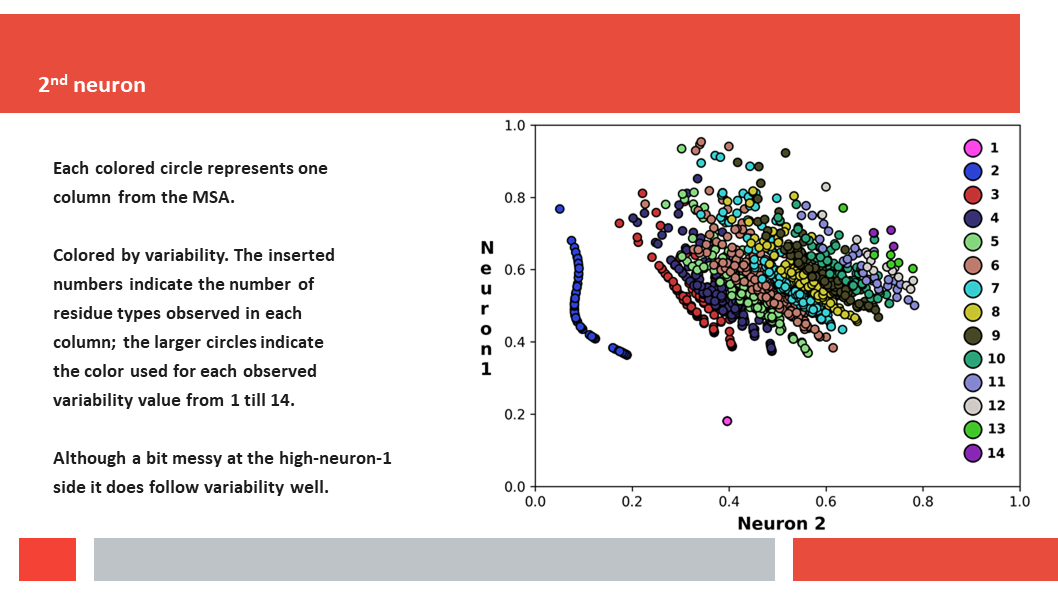
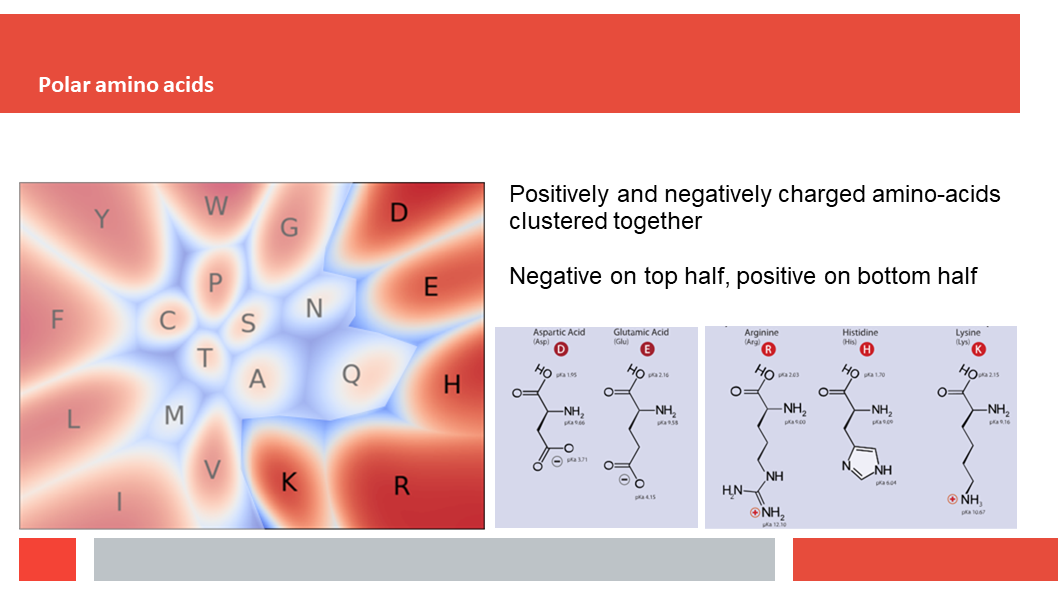
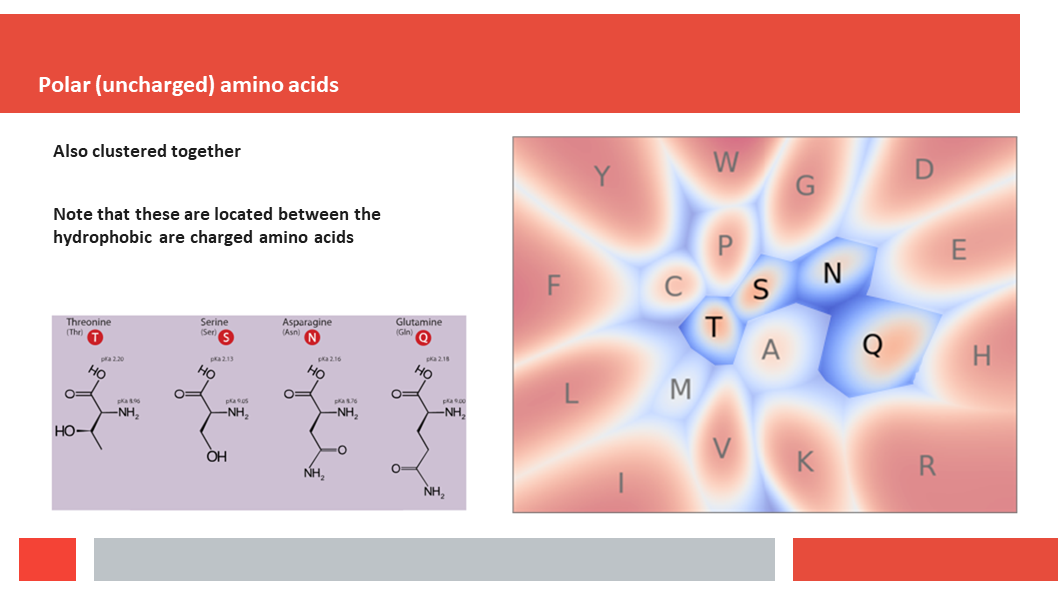
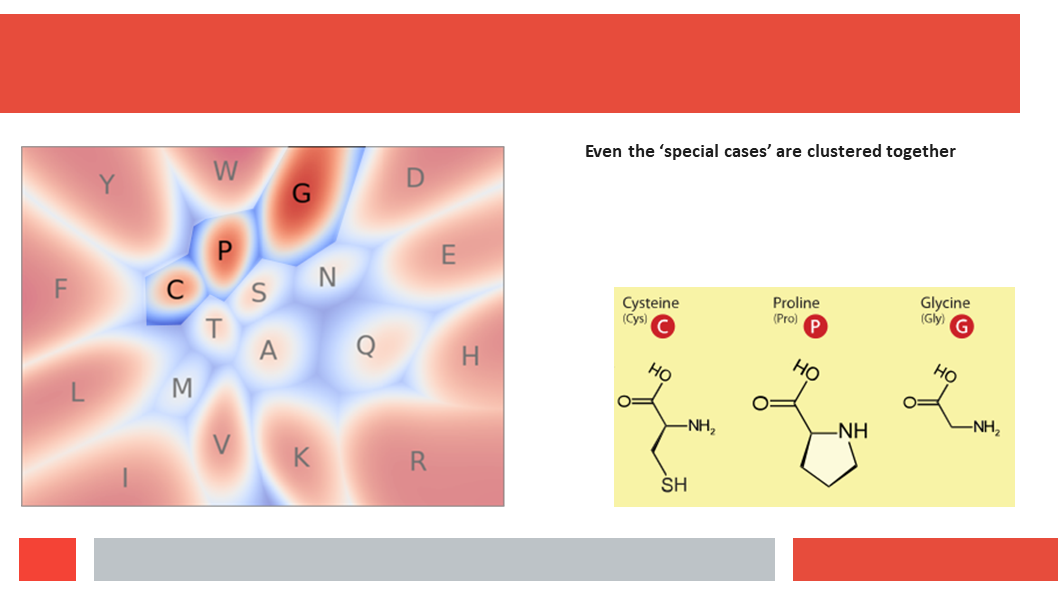
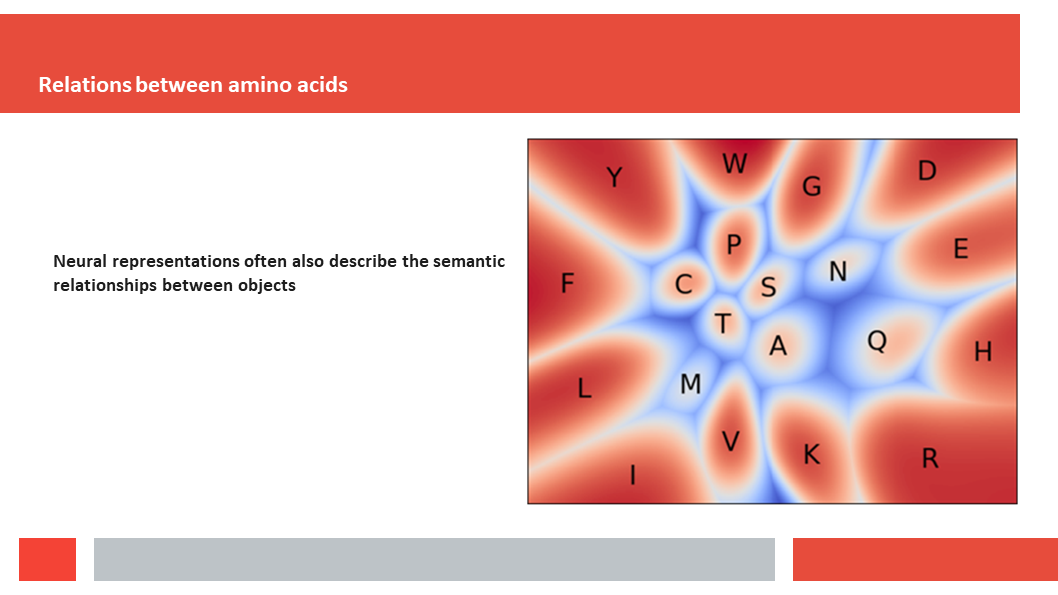
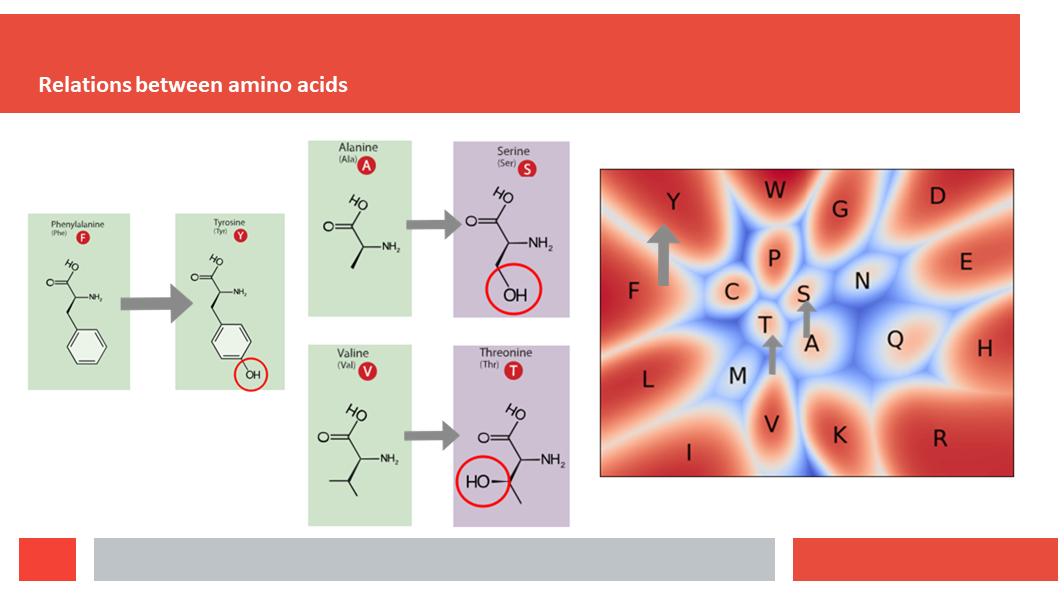
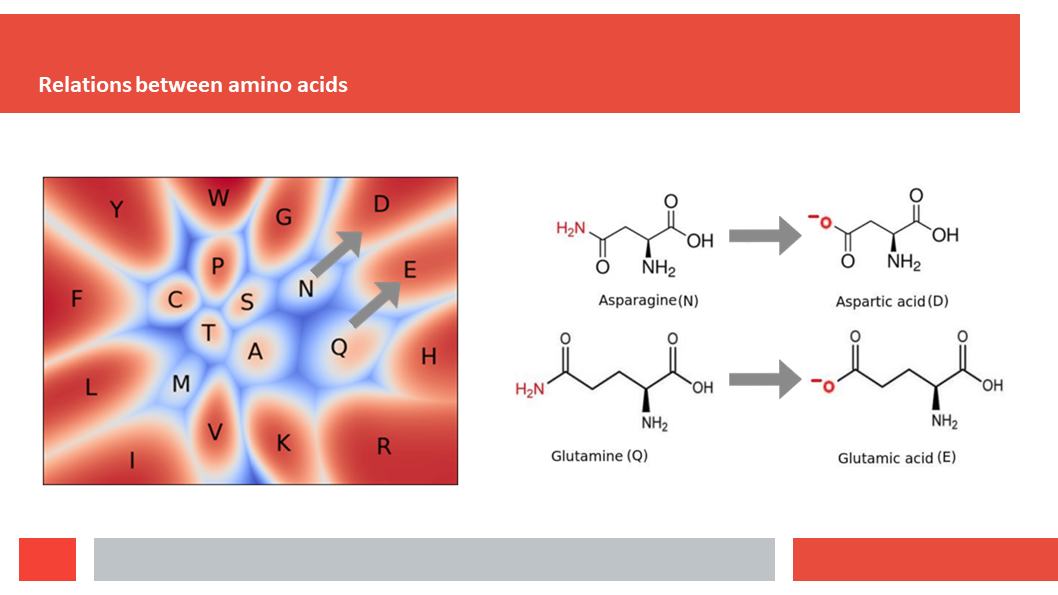
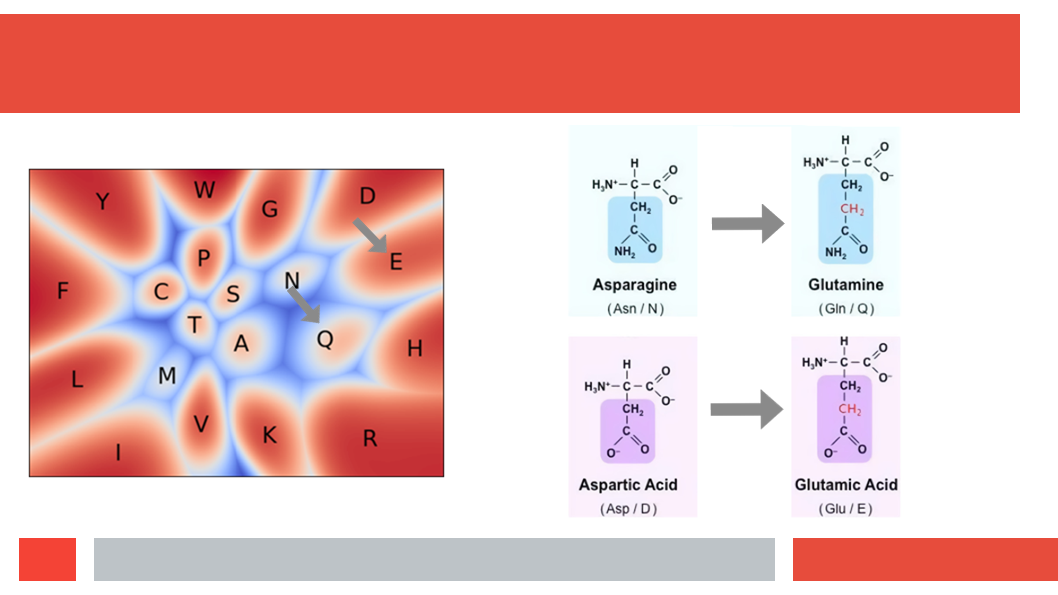
Figure 7. In this article we (Daniel who did the work,
and Gert who talks about it)
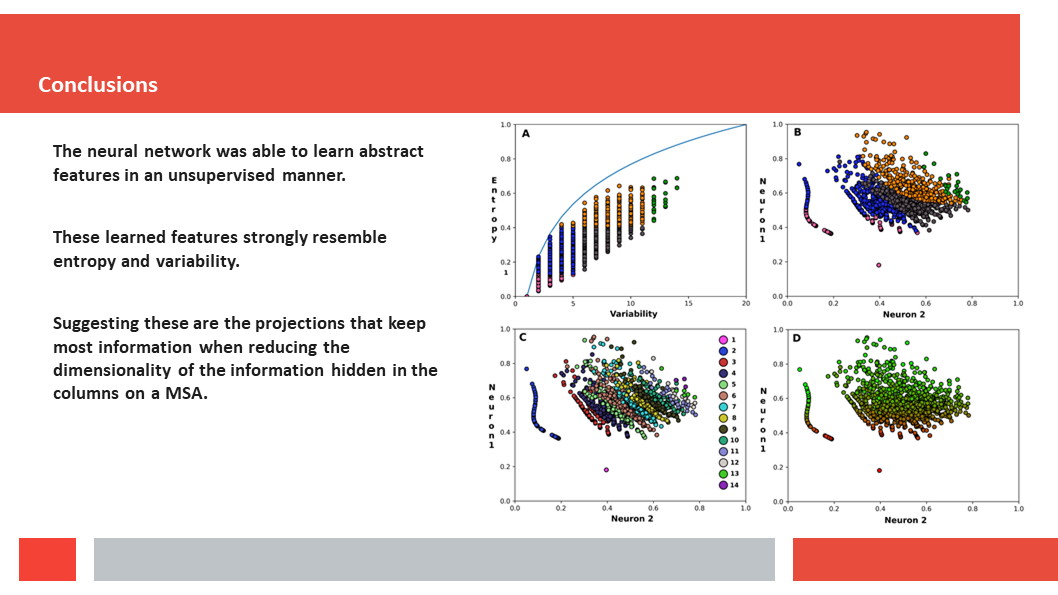
discuss how a deep-learning experiment on HSSP files for the entire human
genome reveals that, just as Laerte Oliveira expected already 20 years ago,
entropy and variability are the two variables that best describe the
variability patterns in columns in MSAs.
(HSSP files are MSAs without insertions. So if the sequence we start
with is N amino acids long, and BLAST finds M homologs for us to align, tha
the final MSA is a matrix of N by M+1; N colums of M+1 amino acids.)
(Click on the image to start this seminar as a movie).