
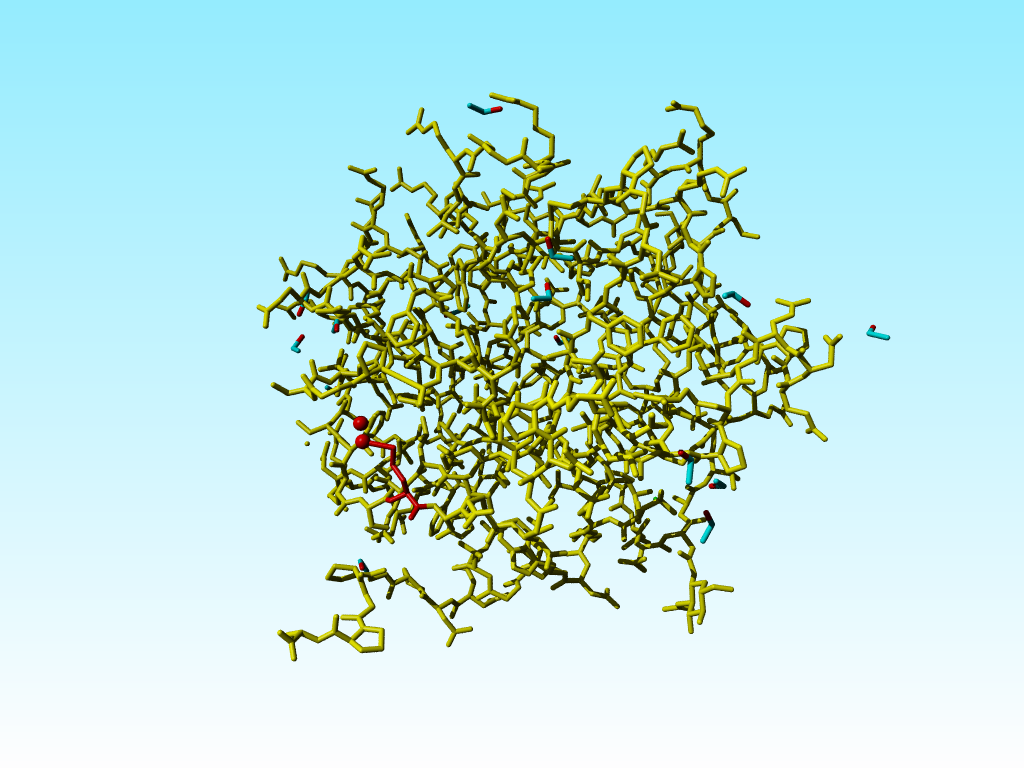
"Glycine" 126 in 1vns. A few residues around it in the sequence are shown in yellow.
JRNL AUTH S.MACEDO-RIBEIRO,W.HEMRIKA,R.RENIRIE,R.WEVER, JRNL AUTH 2 A.MESSERSCHMIDT JRNL TITL X-RAY CRYSTAL STRUCTURES OF ACTIVE SITE MUTANTS OF JRNL TITL 2 THE VANADIUM-CONTAINING CHLOROPEROXIDASE FROM THE JRNL TITL 3 FUNGUS CURVULARIA INAEQUALIS JRNL REF J. BIOL. INORG. CHEM. V. 4 209 1999 |
The file 1vns contains a rather funny Glycine:
ATOM 913 N GLY 126 -9.010 43.791 -20.761 0.00 28.31 N ATOM 914 CA GLY 126 -7.755 44.483 -21.036 0.00 30.13 C ATOM 915 C GLY 126 -7.951 45.840 -21.710 0.00 31.86 C ATOM 916 O GLY 126 -6.995 46.607 -21.859 0.00 31.79 O ATOM 917 CB GLY 126 -6.843 43.600 -21.877 0.00 29.88 C |
Which in 3D looks like:
|
|
"Glycine" 126 in 1vns. A few residues around it in the sequence are shown in yellow. |
Supplemental material
HEADER SERINE ESTERASE 26-MAR-97 1AGY JRNL AUTH A.NICOLAS,C.MARTINEZ,C.CAMBILLAU JRNL TITL THE 1.15 ANGSTROM REFINED STRUCTURE OF FSP CUTINASE JRNL TITL 2 COMPARED TO OTHER MEMBERS OF ALPHA/BETA HYDROLASE JRNL TITL 3 FOLD FAMILY JRNL REF TO BE PUBLISHED |
Another case where a residue became a bit too Alanine is 1agy. In this file the sidchain of Arginine 32 is probably not seen in the density:
REMARK 470 MISSING ATOM REMARK 470 THE FOLLOWING RESIDUES HAVE MISSING ATOMS (M=MODEL NUMBER; REMARK 470 RES=RESIDUE NAME; C=CHAIN IDENTIFIER; SSEQ=SEQUENCE NUMBER; REMARK 470 I=INSERTION CODE): REMARK 470 M RES CSSEQI ATOMS REMARK 470 ARG 32 CG CD NE CZ NH1 NH2 |
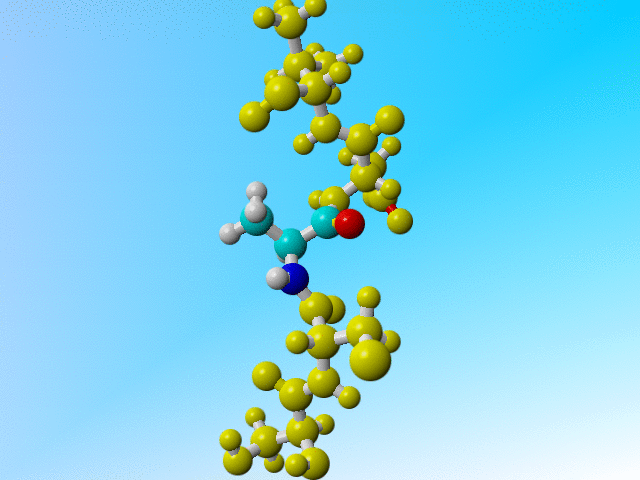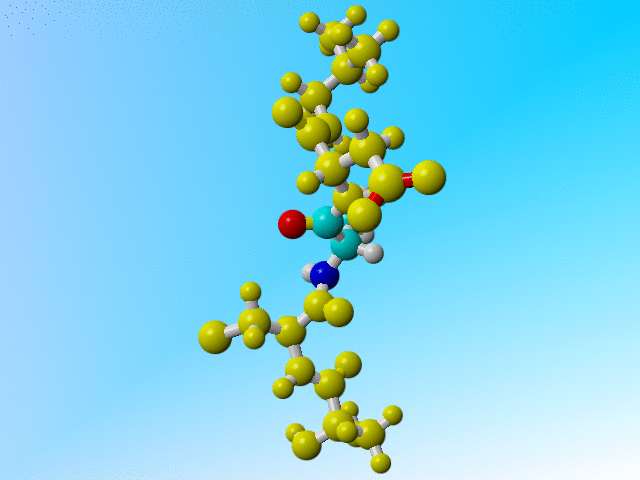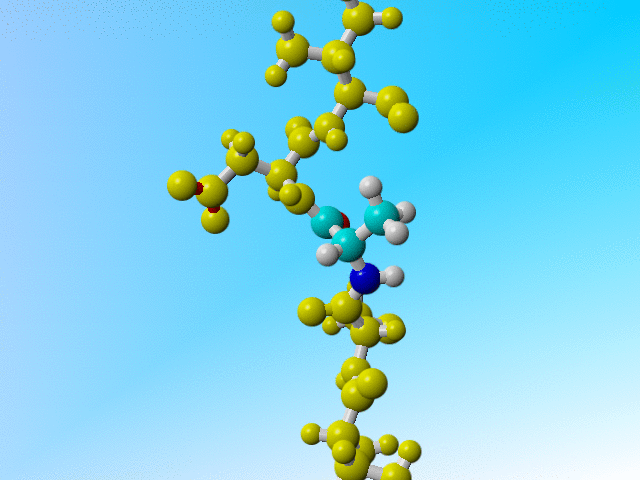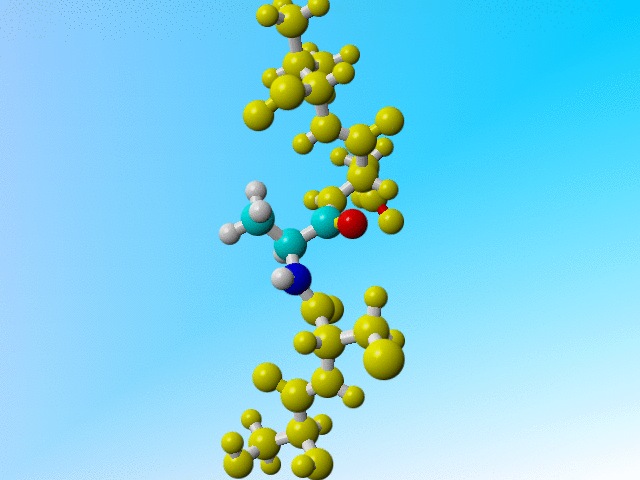
But it is a bit funny to now find three protons on the Cβ of this Arginine:
ATOM 229 N ARG 32 -13.541 61.636 37.307 1.00 11.55 N ATOM 230 CA ARG 32 -13.593 62.560 36.191 1.00 10.28 C ATOM 231 C ARG 32 -14.647 62.070 35.198 1.00 9.94 C ATOM 232 O ARG 32 -14.981 60.886 35.130 1.00 11.25 O ATOM 233 CB ARG 32 -12.225 62.614 35.475 1.00 10.76 C ATOM 234 H ARG 32 -12.759 61.021 37.295 1.00 11.68 H ATOM 235 HA ARG 32 -13.855 63.545 36.555 1.00 10.46 H ATOM 236 1HB ARG 32 -11.843 61.597 35.235 1.00 10.13 H ATOM 237 2HB ARG 32 -11.495 63.119 36.141 1.00 10.21 H ATOM 238 3HB ARG 32 -12.239 63.192 34.527 1.00 10.31 H |
|
Arginine 32 with three hydrogens on its Cβ. |
EU name: 1A7S
(Date: 5 Aug 24 2016 1A7S )
JRNL AUTH S.KARLSEN,L.F.IVERSEN,I.K.LARSEN,H.J.FLODGAARD, JRNL AUTH 2 J.S.KASTRUP JRNL TITL ATOMIC RESOLUTION STRUCTURE OF HUMAN JRNL TITL 2 HBP/CAP37/AZUROCIDIN JRNL REF ACTA CRYSTALLOGR.,SECT.D V. 54 598 1998 |
Sometimes is is nearly unimaginable what legths people are willing to go to really screw up their molecule. Take the file 1a7s. This structure has been solved at 1.1 Ångström resolution:
REMARK 2 REMARK 2 RESOLUTION. 1.12 ANGSTROMS. REMARK 3 REMARK 3 REFINEMENT. REMARK 3 PROGRAM : SHELXL-96 REMARK 3 AUTHORS : G.M.SHELDRICK REMARK 3 REMARK 3 DATA USED IN REFINEMENT. REMARK 3 RESOLUTION RANGE HIGH (ANGSTROMS) : 1.12 REMARK 3 RESOLUTION RANGE LOW (ANGSTROMS) : 15.0 REMARK 3 DATA CUTOFF (SIGMA(F)) : 0.0 REMARK 3 COMPLETENESS FOR RANGE (%) : NULL REMARK 3 CROSS-VALIDATION METHOD : FREE R VALUE REMARK 3 FREE R VALUE TEST SET SELECTION : EVERY 5TH REFLECTION |
Still this file holds many funnies.
Take a look at the validation report to get a list
of many of these funnies.
For example, we know that the Cγ-Sδ and the
Sδ-Cε distances both are close to 1.8 Ångström. In methionine 76 in 1a7s
these distances are: 1.8 and 2.5 respectively (3.2 for the distance from the
Sδ to the B-alternate for the Cε).
By the way, both alternate locations for the Cε have 100% occupancy (labeled
red in the box shown below; in this box the ATOM records for MET-76 in 1a7s are shown).
ATOM 557 N MET 76 -1.650 12.422 22.757 1.00 10.67 N ATOM 558 CA MET 76 -1.094 12.384 24.123 1.00 10.65 C ATOM 559 C MET 76 -1.311 10.990 24.675 1.00 10.32 C ATOM 560 O MET 76 -2.370 10.407 24.465 1.00 12.17 O ATOM 561 CB MET 76 -1.951 13.375 25.005 1.00 13.71 C ATOM 562 CG MET 76 -1.735 14.859 24.519 1.00 16.65 C ATOM 563 SD MET 76 -2.320 15.970 25.782 1.00 41.53 S ATOM 564 CE AMET 76 -3.232 17.467 24.022 1.00 45.15 C ATOM 565 CE BMET 76 -3.402 18.790 24.744 1.00 43.54 C |
This error that alternate atoms have all full occupancy occurs many times in 1a7s. I would not be surprised if this was the cause for the very funny bond-lengths. The cause could also lie in the look 44-47 for which the density is missing (at least the atoms are missing, so I assume this to be the result of poor density). Perhaps the methionine side chain got modelled in some density for some residue in the 44-47 loop.
Supplemental material
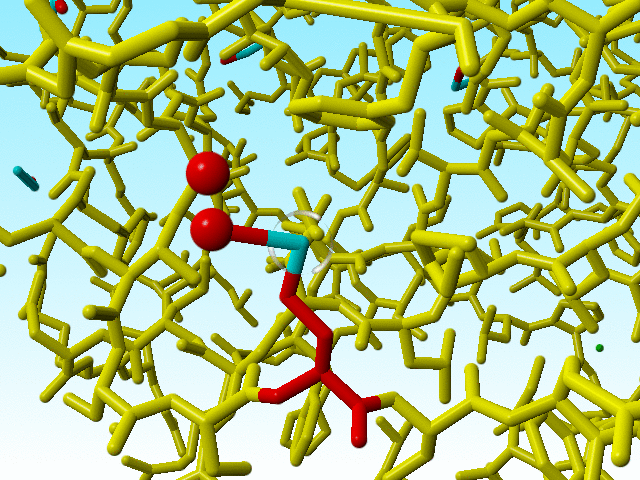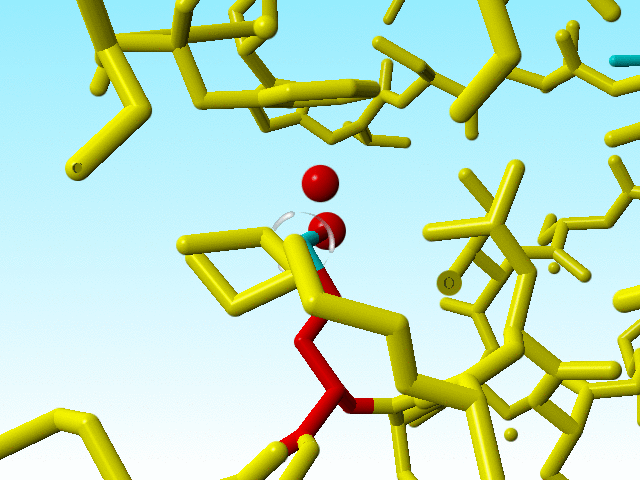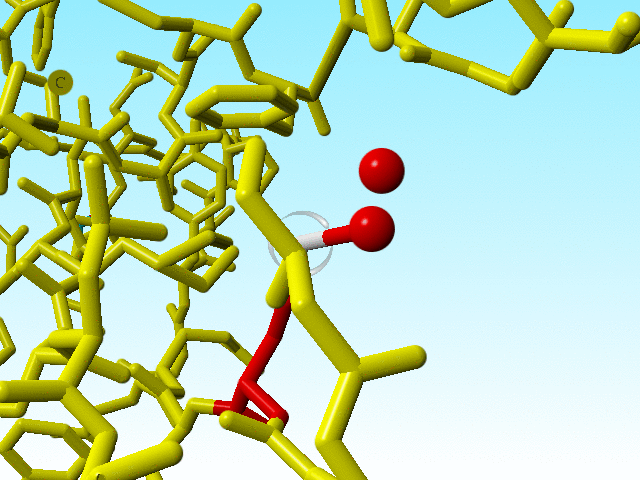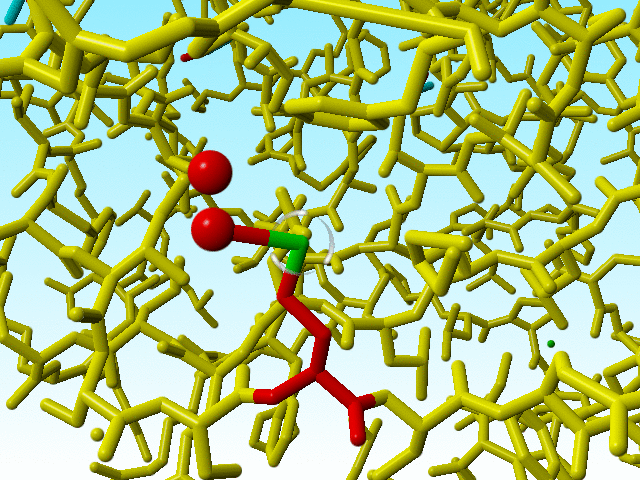
|
Methionine 76 in 1a7s in red. The alternate atom Cε-B is so far away from its covalent neighbour Sδ that I cannot get this software to draw that bond. |
|
Blow up of methionine 76 in 1a7s as shown just above. |
These errors in methionine 76 are therefore so "interesting" because there are two other methionines immediately adjacent to methionine 76 that are OK (in terms of bond lengths). The one (methionine 91) has alternate atom positions for its Sδ and Cε, so it is possible to get it right with the wrong occupancies.
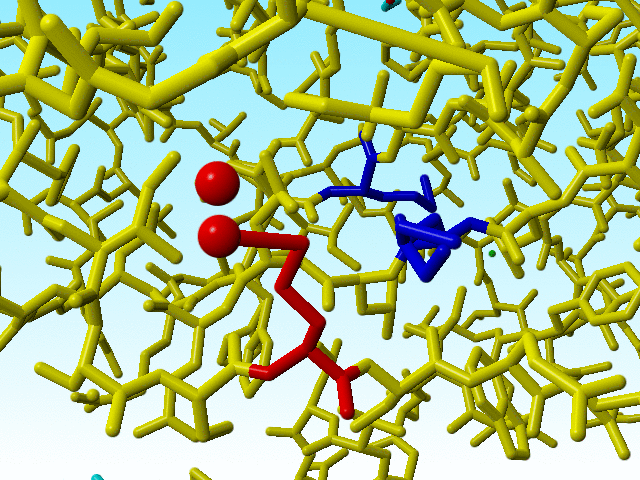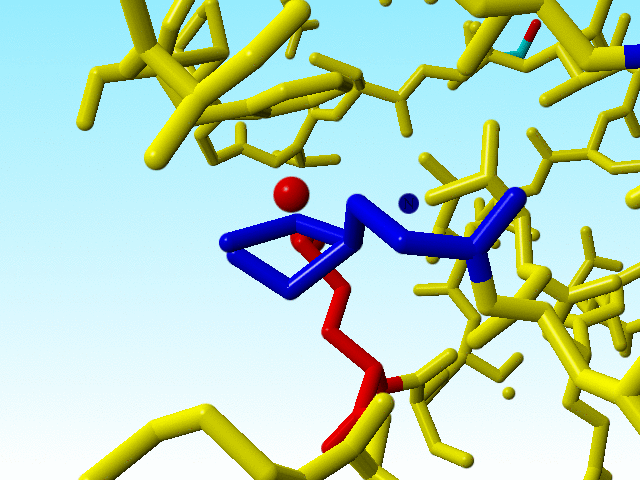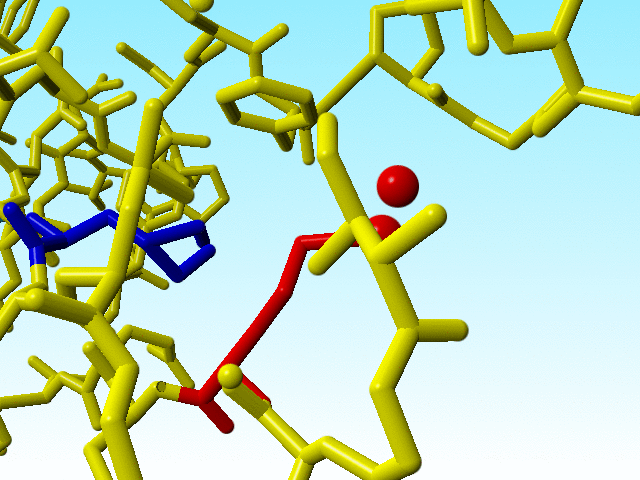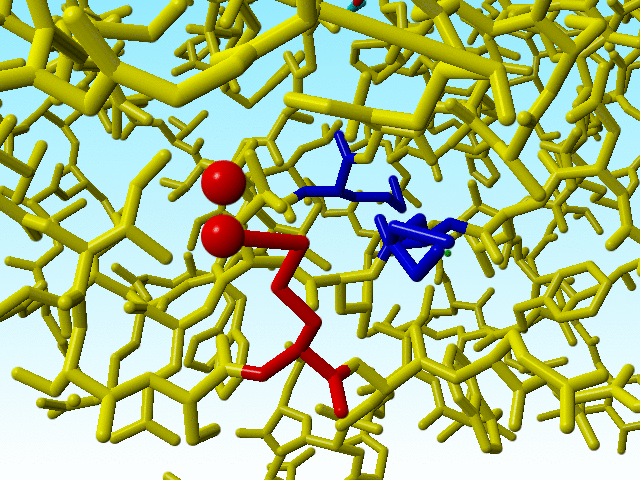
|
The two methionines that are close to the funny methionine 76 are shown in blue. |
It is commonly known that the guanidinium group Cδ-Nε-Cζ-Nη1(Nη2) in arginines must be flat. Sometimes there is doubt about how flat it should be, but the Nε-Cζ-Nη1(Nη2) atoms all have SP2 hybridisation. In most arginines in 1a7s this chemical knowledge has been applied, but not in arginine 166, in which the Nε is SP3.
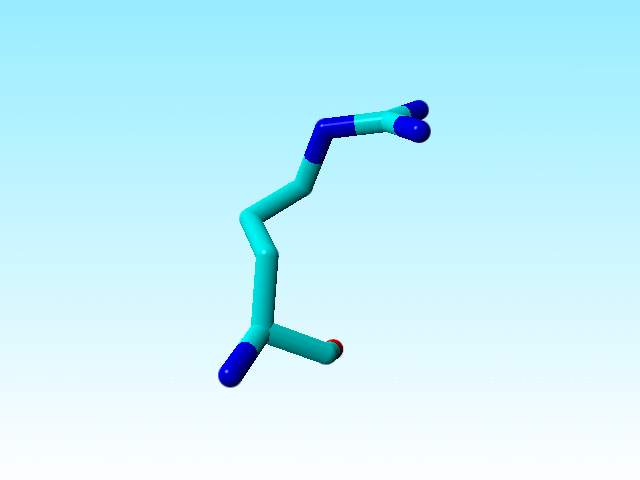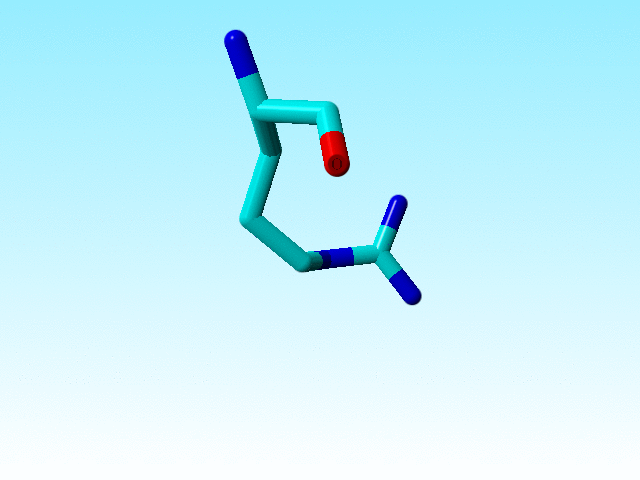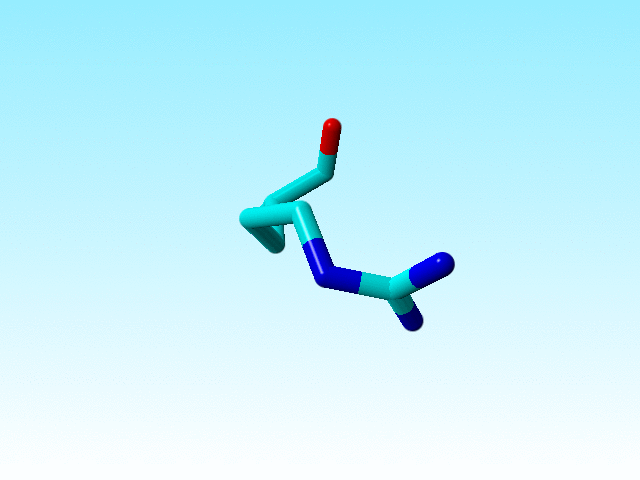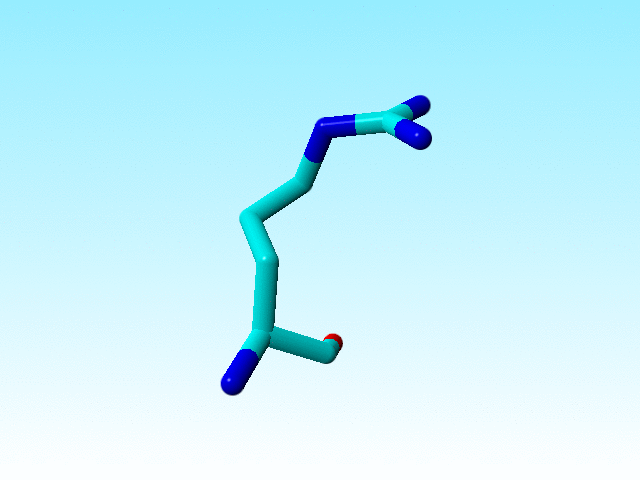
|
Arginine 166 in 1a7s has an SP3-hybridized Nε. |
The arginines 63 and 65 form hydrogenbonds between their positively charged side chain nitogens (with short distances, even for a normal hydrogenbond). I know that these arginines have slightly higher B-factors, but the B-factors are not so extremely high that the density can be considered absent.
Supplemental materialThese arginines make symmetry contacts with more likely hydrogenbonding partners, so it is possible that they swapped density with those residues in a symmetry related molecule.

|
The two arginines that make hydrogen bondins are indicated. Residues in symmetry related molecules that fall within 5 Ångström of any atom in the molecule in which the two arginines are shown are displayed in brown. |
There are many memory aids for figuring out where to stick the Cβ relative to the backbone N-Cα-C atoms. The CORN law is one of the better known ones:
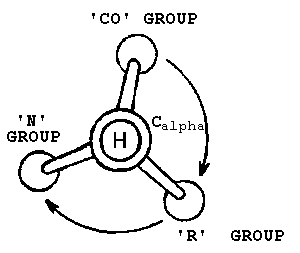
|
The normal L configuration can be remembered by the CORN law. Imagine looking along the H-Cα bond with the H atom closest to you. When read clockwise, the groups attached to the Cα spell the word CORN. |
Sometimes this goes wrong, and WHAT_CHECK warns for the existence of D amino acids (remember that D amino acids don't need to be wrong, but you better check them anyway). Valine 50 in 1a7s is a bit funny in that it is neither an L amino acid, nor a D amino acid, but it falls just half-way inbetween.
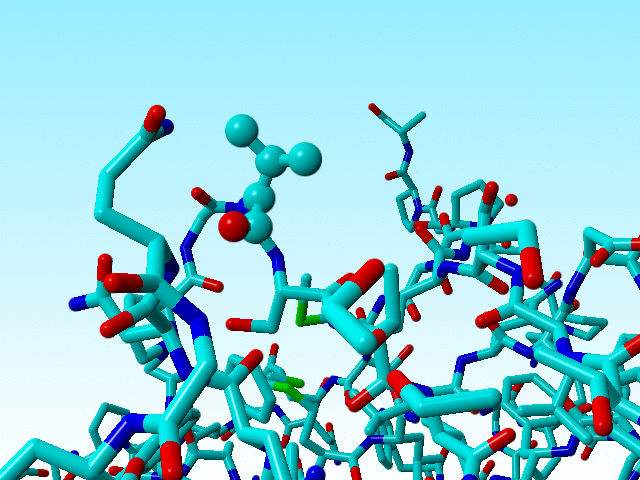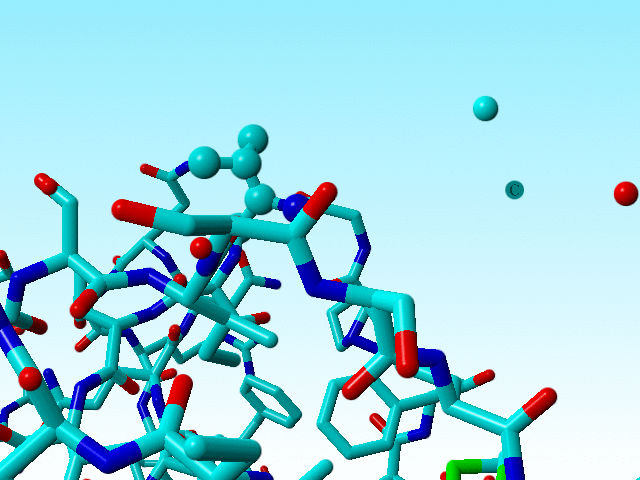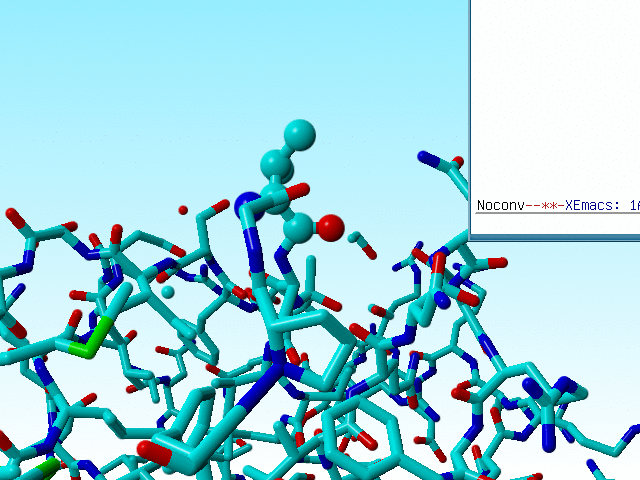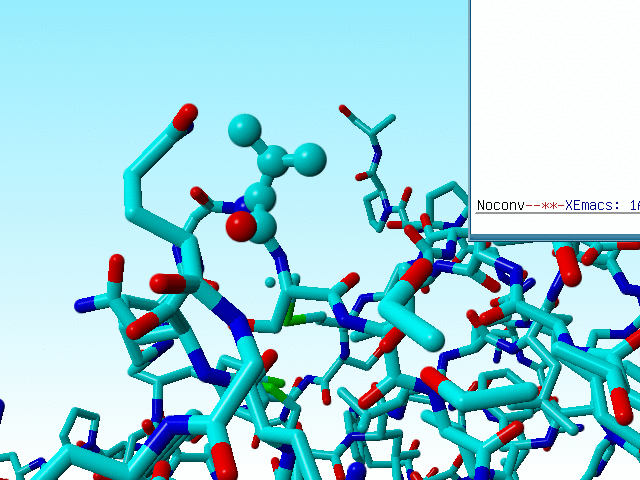
|
The three atoms connected to the Cα of valine 50 in 1a7s have a geometry that suggests that the Cα is SP2 hybridized, but the bond lengths are within experimental error in agreement with SP3. |
EU name: 406D
(Date: Aug 24 2016 406D )
JRNL AUTH S.A.SHAH,A.T.BRUNGER JRNL TITL THE 1.8 A CRYSTAL STRUCTURE OF A STATICALLY JRNL TITL 2 DISORDERED 17 BASE-PAIR RNA DUPLEX: PRINCIPLES OF JRNL TITL 3 RNA CRYSTAL PACKING AND ITS EFFECT ON NUCLEIC ACID JRNL TITL 4 STRUCTURE JRNL REF J.MOL.BIOL. V. 285 1577 1999 |
This structure was solved by the main author of the most-used X-ray refinement program (XPLOR), Axel Brunger. The XPLOR software was written with more emphasis on userfriendlyness than on scientific rigor.
This structure has seen one of the weirdest refinement procedures I know about. The occypancies of all atoms vary widely between 0.84 and 1.17...
The placement of waters in this file is also funny to say the least:

|
The RNA in yellow and the waters in red in 406d. |
The long rows of water still make sense as can be seen when I add symmetry related molecules:
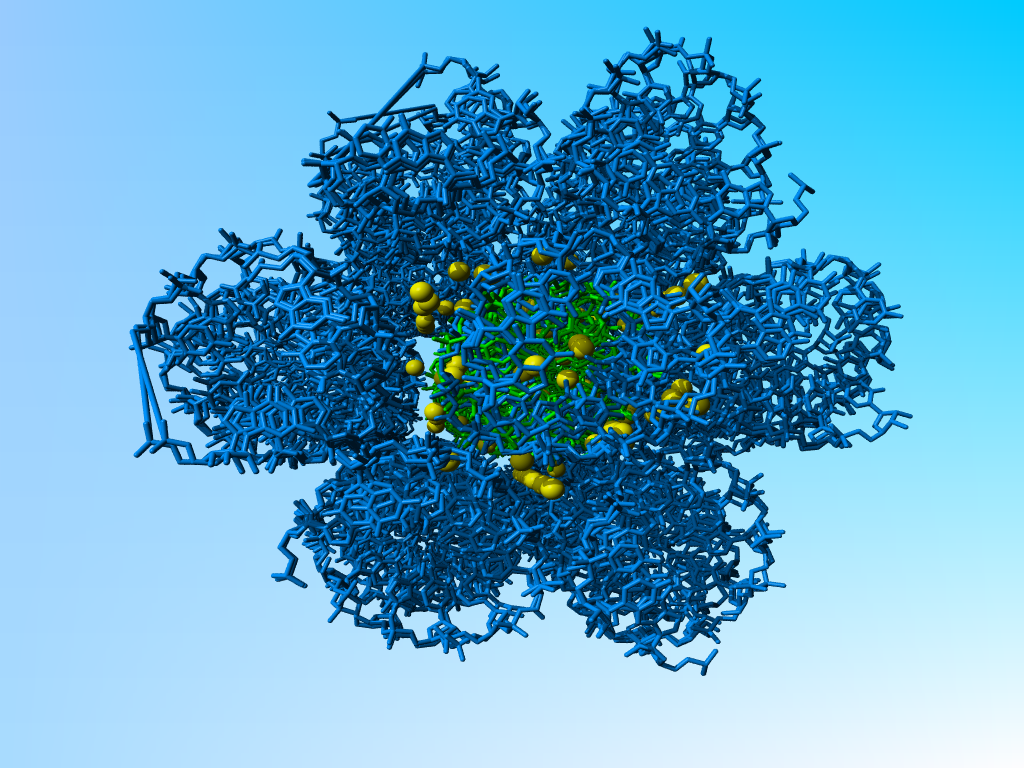
|
406d (in green) with a layer of symmetry related molecules (in blue) around it. The waters are in yellow. The funny rows of warers are found in the tunnels formed when three molecules of RNA come together. |

|
Enlargement of the previous picture, focusing on a funny row of waters. The funny long lines are the result of the incompleteness of the symmetry related molecules (in blue) far away from the central (green) one. |

|
Why there are waters only at one side of the molecule remains a miracle to me. |
Also, I can understand the principle of heterogeneous RNA samples (I cannot understand why one wants to crystallize that in such an artificial form, but that is a bio-scientific question that is unrelated to X-ray structure refinement). I can also understand that one doesn't want to encode heterogeneity in the usual way when it is as massive as in this case. But I don't understand how one can properly refine a structure when at most positions there are atoms with nearly 1.0, 1.0, or even more than 1.0 occupancy.
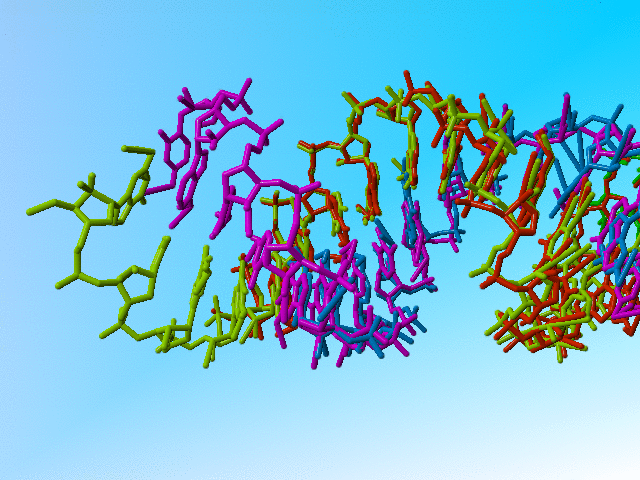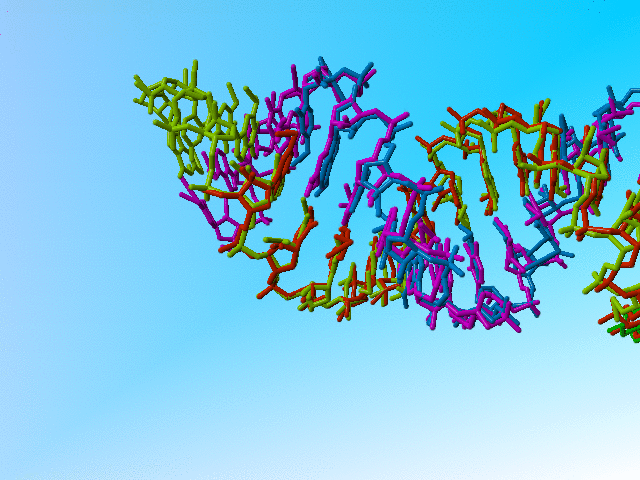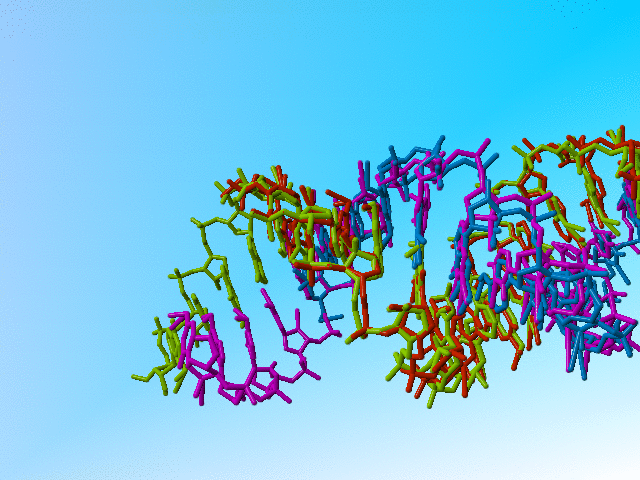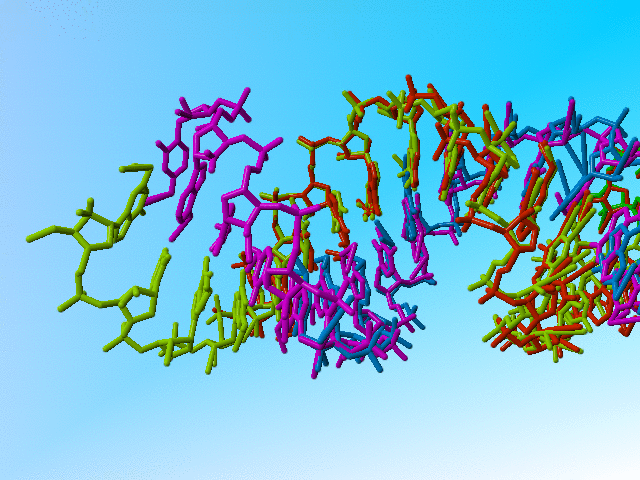
|
Full occupancy superposed molecules in 406d |
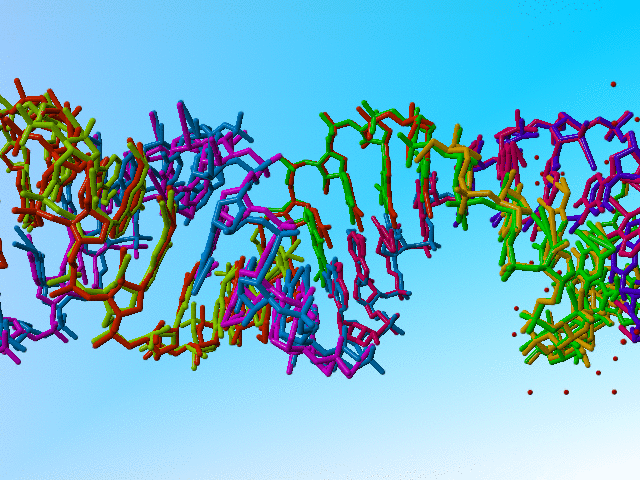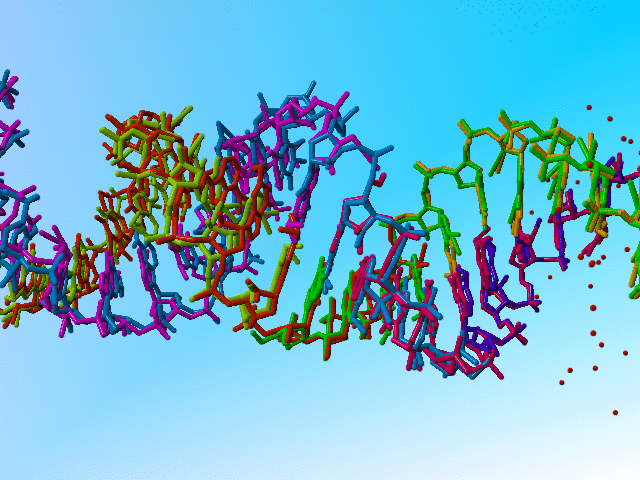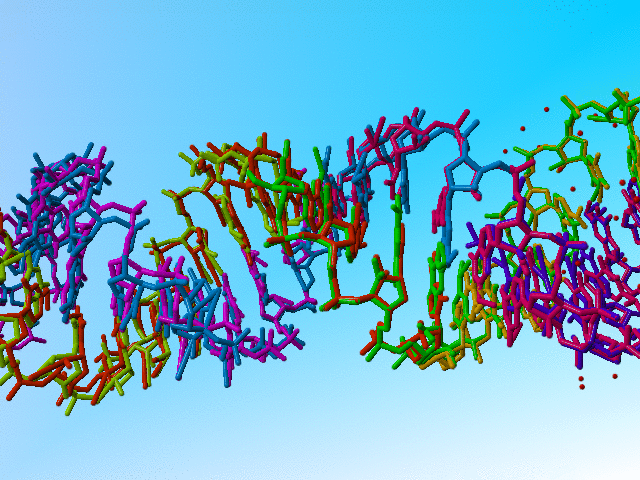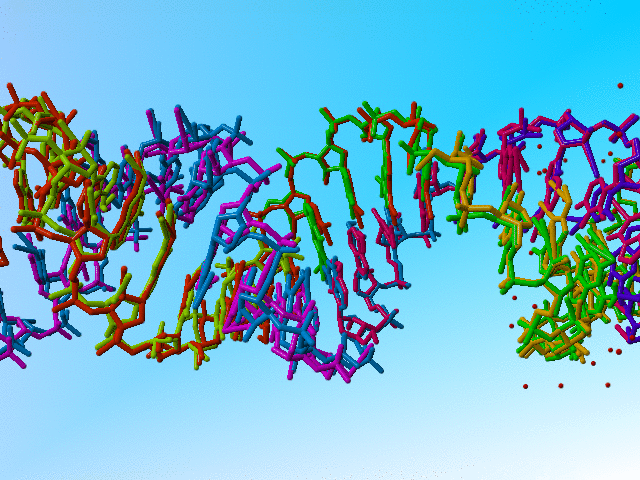
|
Full occupancy superposed molecules in 406d |
Ps. The funny bonds are made by YASARA when it gets confused about which of the
overlapping atoms belongs to which residue. The admininstration of a file
written by XPLOR/CNS in the lab of the author of XPLOR/CNS is such a mess that
YASARA cannot sort it out.
EU name: 1F8H
(Date: Aug 24 2016 1F8H )
JRNL AUTH T.DE BEER,A.N.HOOFNAGLE,J.L.ENMON,R.C.BOWERS, JRNL AUTH 2 M.YAMABHAI,B.K.KAY,M.OVERDUIN JRNL TITL MOLECULAR MECHANISM OF NPF RECOGNITION BY EH JRNL TITL 2 DOMAINS JRNL REF NAT.STRUCT.BIOL. V. 7 1018 2000 JRNL REFN ASTM NSBIEW US ISSN 1072-8368 |
This is an NMR structure. Nothing (really) wrong with that. But browsing through the coordinates I see a series of prolines at position 6 that all look more or less like:
ATOM 1 N PRO A 6 0.096 5.633 -27.811 1.00 0.00 N ATOM 2 CA PRO A 6 -1.167 5.923 -27.086 1.00 0.00 C ATOM 3 C PRO A 6 -1.059 5.487 -25.622 1.00 0.00 C ATOM 4 O PRO A 6 -0.019 5.055 -25.167 1.00 0.00 O ATOM 5 CB PRO A 6 -2.203 5.079 -27.821 1.00 0.00 C ATOM 6 CG PRO A 6 -1.428 3.958 -28.442 1.00 0.00 C ATOM 7 CD PRO A 6 -0.023 4.453 -28.676 1.00 0.00 C ATOM 8 HA PRO A 6 -1.421 6.968 -27.160 1.00 0.00 H ATOM 9 1HB PRO A 6 -2.934 4.695 -27.123 1.00 0.00 H ATOM 10 2HB PRO A 6 -2.685 5.663 -28.589 1.00 0.00 H ATOM 11 1HG PRO A 6 -1.414 3.108 -27.772 1.00 0.00 H ATOM 12 2HG PRO A 6 -1.876 3.677 -29.382 1.00 0.00 H ATOM 13 1HD PRO A 6 0.696 3.697 -28.389 1.00 0.00 H ATOM 14 2HD PRO A 6 0.113 4.735 -29.708 1.00 0.00 H |
But then, in model 4:
ATOM 1 N PRO A 6 0.580 2.000 2.000 9.00 0.00 N ATOM 2 CA PRO A 6 -0.716 4.851 -26.695 1.00 0.00 C ATOM 3 C PRO A 6 -0.861 4.486 -25.217 1.00 0.00 C ATOM 4 O PRO A 6 0.016 3.880 -24.633 1.00 0.00 O ATOM 5 CB PRO A 6 -1.745 4.115 -27.548 1.00 0.00 C ATOM 6 CG PRO A 6 -1.049 2.872 -28.007 1.00 0.00 C ATOM 7 CD PRO A 6 0.419 3.201 -28.104 1.00 0.00 C ATOM 8 HA PRO A 6 -0.802 5.917 -26.840 1.00 0.00 H ATOM 9 1HB PRO A 6 -2.613 3.866 -26.953 1.00 0.00 H ATOM 10 2HB PRO A 6 -2.028 4.714 -28.398 1.00 0.00 H ATOM 11 1HG PRO A 6 -1.206 2.077 -27.291 1.00 0.00 H ATOM 12 2HG PRO A 6 -1.419 2.577 -28.976 1.00 0.00 H ATOM 13 1HD PRO A 6 0.682 3.474 -29.114 1.00 0.00 H ATOM 14 2HD PRO A 6 1.017 2.365 -27.765 1.00 0.00 H |
I am really curious about the nine red nitrogens.