We will now go through the different parts of a WHAT IF check report, discussing the different points. The report we are following is an old version of the 103L. This is in general a fairly good structure, so sometimes we will look at some bad examples too. If you want, you can have a look at the most recent version of the check report of 103L, or browse the sections used here quickly through an index.
For the example, see "Nomenclature".
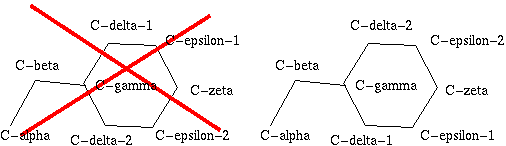
Some of the 20 amino acid types in proteins have atoms that "look" the
same. So, for instance, the C-delta-1 and C-delta-2 atoms in
phenylalanine look the same, and thus all PHE residues would have two
ways of naming the atoms:
This would result in infinite confusion. So, a committee designed a standard: the torsion angle Chi-2 (defined by C-alpha, C-beta, C-gamma, C-delta-1) should always be between -90 and 90 degrees.
Most of the time this convention works, and so WHAT IF enforces it, and will give a warning about non-compliant cases. There are two problems:
Similar considerations hold for ASP, GLU, PHE and TYR.
A slightly different problem exists for VAL, THR, ILE, LEU and ARG. Here the two atoms that look similar in a flat drawing of the residue are actually very different in 3-D. This makes it easier to define the conventions. And it makes it really bad if one of these shows up as an error (ARG is a slightly weird case, since most programs do not seem to know that there is a difference between the two terminal NH2 groups).
For the example, see "Other administrative checks"
If there is more than one chain in the structure (E.g. the structure of Triose phosphate isomerase consists of 2 chains that are identical in sequence), the PDB file contains a unique chain identifier (a single letter) for each one. This is useful to distinguish real two-chain cases from cases where the crystallographers do not give coordinates for one or more residues in the middle of a chain (e.g. because the loop is not well defined).
The chain name check in WHAT IF gives an error message if there is more than one chain with the same name.
Not all atoms in a structure are always at the same position. That is in protein structures especially true for water molecules, but in accurate structures also for protein atoms (one can observe different locations of the same side chain).
In the PDB file each atom has a "Weight": a number between 0 and 1 that specifies what fraction of the time the named atom spends in that position.
The last option might seem useless, but actually some crystallographers like to specify atoms that way if they can't see where they should be. The coordinates are in such cases a reasonable guess, but there was no data to support it.
The weight check verifies that all numbers specified are between 0 and 1 (inclusive).
Impact on modelling by homology: None. But there is a related problem with structures that contain atoms with 0.0 weight: Since there is no data supporting these atom positions, these have to be treated as modelled already. And that modelling might not have been done in a very intelligent way!
Not all crystallographers do as described above: give all atoms they know should be there, just set the WEIGHT to 0.0 if you can't see them. Actually most crystallographers just leave out all atoms they can't see.
The check for missing atoms will alert you if there are any amino acids that are not complete.
Impact on modelling by homology: Residues that are incomplete need to be modelled into place. This increases the complexity of the modelling process, and hence could decrease the accuracy of the result.
A special case is the C-terminal oxygen. The peptide bond is formed from an acid (COOH) group and an amine (NH2) group. The peptide group becomes "CO-NH", ejecting one O and two H's as water (H2O).
The last residue in each chain should end in a COOH group, adding one oxygen to the normal residue.
The C-terminal oxygen check verifies whether all chains have exactly one of these C-terminal oxygens at the end.
Most of the symmetry checks in WHAT IF are described in J. Appl. Cryst. (1994) Vol 27 pp 1006--1009.
For the example see "Symmetry"
Many of the parameters that describe the symmetry of the structure are given twice in the header of the PDB file: once in the CRYST1 card, and once in the SCALE cards. The two representations are very different, and so can trigger independent types of mistakes.
WHAT IF verifies the consistency of the information on the CRYST1 and SCALE cards, and if they differ, will try to select the most reliable of the two based on the kind of discrepancy encountered. In most cases a corrected version of the information will be printed as well.
Among the many different kinds of problems detected here are:
Impact on modelling by homology: None.
The International Tables for X-ray crystallography (Kynoch press, Birmingham) list a number of conventions for unit cells. If no attention is payed to these conventions, an multitude of unit cells can be used to describe the same lattice. Comparing different unit cells would be absolutely meaningless. The conventions make sure the unit cell specified is the "most rectangular" cell with the "shortest axes".
WHAT IF will use a procedure described by Y. Le Page (J. Appl. Cryst. (1982) Vol 15 pp 255--259) to derive the standard unit cell, and complain if this looks different from the specified cell.
Impact on modelling by homology: None.
Brian Matthews (J. Mol. Biol. (1968) Vol 33 pp 491--497) has described a parameter that is related to the solvent content of the protein structure. This so-called Matthews' coefficient should lie between 1.5 and 4.0 for most structures. WHAT IF checks this value. In most cases where the value appears to be out of range, the value of "Z" on the CRYST1 card (the number of identical protein chains in the unit cell) is incorrect.
Impact on modelling by homology: None.
Impact for crystallographer: Check the rules for the calculation of Z in the PDB manuals.
A side-effect of the cell reduction process described above is that super-cells with higher lattice symmetry are found. However: higher lattice symmetry could be pseudo-symmetry. Or it could be not supported at all by the structure.
In cases where there are independent molecules, WHAT IF will calculate superposition transformations in the frame of the standard unit cell. If the resulting transformation looks somewhat like a symmetry transformation (lots of 1.0 and 0.0 values), a message will be produced that asks the crystallographer to check this.
Impact on modelling by homology: If two "identical" molecules are independent, this gives two starting points for modelling. If, however, WHAT IF is correct in its assumption that the two molecules should be identical, a superposition of the two will give an impression of the magnitude of the uncertainty in the crystal structure determination.
Impact for crystallographer: The best way to check whether any higher symmetry is real would be to:
For pairs of "identical molecules" that are independent in the asymmetric unit, WHAT IF will produce a pair of plots (the number of plots is limited to prevent hundreds of pages of checking reports for 11-mers):
Plots are only produced if they contain potentially interesting information.
Normally, structures of identical sequence should be quite similar. Any large differences in the structure as apparent from the plots are indicative of potentially very interesting areas in the structure (if the difference is localized), or of a problem in the refinement (if there are differences everywhere).
Impact on modelling by homology: A structure with two independent molecules with local differences could be used to get an indication of the functionality of a protein. If there are global differences, their extent can give an impression of the reliability of the structure.
Impact for crystallographer: In most cases structures should be refined with non-crystallographic symmetry restraints or constraints. The affected structural region should be allowed to refine freely in both molecules only if there are very clear indications that the conformers are different.
For the example, see "Chirality". Another (worse) example can be found in the report for "104L"
The chirality check in WHAT IF does more than just check the D- or L-ness of the amino acids making up the protein. For each of the atoms in the protein chain that have 3 bonds to non-hydrogen atoms, the configuration of those connections is verified.
This is done using the "improper dihedral" consisting of the atom itself and the 3 connecting atoms:
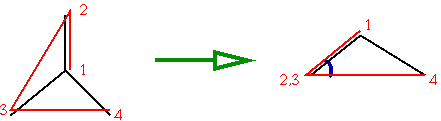
The "improper dihedral" in this case is indicated by the red lines, and its value can be seen by looking along the line from 2 to 3 (indicated by the blue arc in the right picture): in this case the value is around -35 degrees. Values of +35 and -35 are normal for non-planar configuations, 0 degrees will be obtained if the configuration is planar.
For each of the atom types in proteins, the mean value and standard deviation for the corresponding improper dihedral is calculated from the WHAT IF database of reliable structures. These are used to calculate chirality Z-scores for all atoms except planar side-chain atoms (these are treated separately in a planarity check). The outliers will be reported in a table.
The four outliers reported in the table in the example are C-alpha's for which the improper dihedral is too close to 0. This means that the "umbrella" defined by the configuration around these C-alpha atoms is "too flat". In 104l there are many more of these kind of problems, and also two planarity problems in the backbone.
Impact on modelling by homology: Small errors like in 103L are normally easily fixed by geometry regularization, but they might indicate that there are improperly placed side-chains in the neighbourhood that force a residue into a strange conformation. Any inverted chirality is either an indication of non-protein residues or of severe problems in the structure.
Impact for crystallographer: Chirality deviations might be indication of misplaced side chains. It might be good to check the electron-density map around the problem to find out.
For the example, see "Bond lengths". A bit worse in the report for "104L"
Bond lengths are quite well-known entities. Since similar bonds have similar lengths, and there are plenty of highly accurate small molecule structures that contain small peptides, there is a very good standard for bond lengths in proteins. This includes averages and standard deviations for all different bond types (R. Engh and R. Huber, Acta Crystallogr. vol A47, pp 392-400). These standards are used by WHAT IF to calculate Z-scores for each of the bonds in the structure. All outliers (Z<-4 or Z>4) are listed in a table. Since there are many bonds in a protein, it is normal to have 1 or 2 entries in the table; it might be worth checking them anyway. Any systematic deviation, however, is cause of serious concern. An RMS Z-score is also calculated, indicating whether the overall distribution of bond-lengths is normal.
The impact of a too-tight constraint on bond lengths, resulting in an RMS Z-score below 1, is not severe. It is definitely not a problem for low through mid-resolution X-ray structures or NMR structures. It might not even be serious for atomic resolution structures. (See e.g. the check report of the very good structure 1CTJ).
There is one other thing that is checked for crystal structures: For all deviations from the ideal bond lengths for all bonds in the structure, is there a preferential direction? e.g. are most bond lenghts along the "a" axis too short, and along the "b" axis too long? If this occurs, it is taken to be an indication of errors in the determination of the crystallographic unit cell. Since the determination of the unit-cell normally does not get a lot of attention, this problem occurs in quite a number of cases! Especially synchrotron data collections are very sensitive here (J. Appl. Cryst (1986) Vol 19 pp 134--139). It is essential to check the cell dimensions before attempting Molecular Replacement techniques! For an example see the report for "104L". This directional bondlength check will only be executed when the PDB file contains at least some protein. This because DNA has naturally already a high directionality that can easily convert a random error into a systematic one.
Impact on modelling by homology: None. Any bond length problems are of <0.1 Angstrom scale, much too small too be concerned about. However, since it is very simple to do correctly, it might indicate that either you're dealing with an old structure, or the crystallographer has not put much time in it (and this might have caused other, more significant, problems).
Impact for crystallographer: Individual problems are indicative of refinement problems, and warrant an extra look at the map.
For the example, see "Bond angles"
Like for the bond lengths, mean values and standard deviations of bond angles can be obtained from small molecule structures. These can be used to calculate Z-scores for all bond angles, and an RMS Z-score for the whole protein.
It seems that bond angles are more difficult to refine than bond distances; so many structures show a number of outliers. I personally think this is reason for concern, but there is not much we can do. One other effect of this is that normal RMS Z-scores are not 1.0, but closer to 1.5. So WHAT IF will only start complaining if it is larger than 2.0.
Impact on modelling by homology: Just a little bit more than for the bond lengths, because we're talking about slightly larger displacement of the atoms.
Impact for crystallographer: Individual problems are indicative of refinement problems, and warrant an extra look at the map.
For the example, see "Torsion angles" and "Ramachandran Plot".
Torsion angles are much more difficult to verify than either bond lengths or bond angles. This is because they are not restrained near a single "right" value, with deviating values being "wrong".
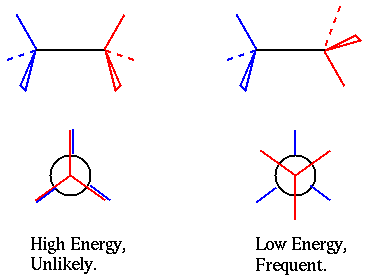
Instead, most torsion angles have three different "good" values (red and blue atoms being as far from each other as possible, also named "staggered") and three different "wrong" values (red and blue atoms being in fairly close contact, also named "eclipsed"). Because of this difficulty, and also because of the fundamental differences between the different torsion angles in a protein, a number of different checks are executed by WHAT IF.
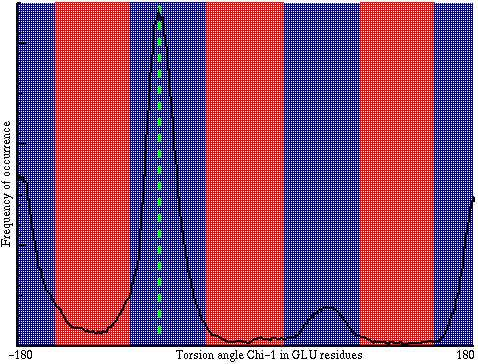
In the plot you can see the effect of the 3 favourable and 3 unfavourable values of the torsion angle. If the value of the torsion angle observed in a protein occurs very frequently in the database (e.g. the green line), that torsion angle gets a good score. If it is very infrequent (mostly in the red areas) it gets a low score. The average score for all torsion angles in the residue excluding omega (so including "phi", "psi" and all "chi" angles) is now used to score the residue. These scores are calculated like Z-scores, but because the underlying distributions are not "normal distributions", the resulting properties are slightly different. Important to know: if the value is below -2.0 the residue feels "strange", if the value is below -3.0 the residue feels "wrong" (Take a look in the report for "104L").
Impact on modelling by homology: A few of these might not be a disaster, but a lot of low scoring residues are an indication of mis-placed side chains. This definitely has an adverse effect on modelling work.
Impact for crystallographer: Individual problems are indicative of misplaced or disordered residues, and warrant an extra look at the map.
The Phi/Psi plot (Ramachandran plot) was the first serious verification tool available for protein strucures. It is based on the fact that backbone torsion angles phi and psi are rather restricted in their possibilities because there must be a place for the side chains of the residues. The "areas" in the Ramachandran plot have clear "alpha-helix"-like and "beta-strand"-like areas, and some allowed loop conformations separate from those. The idea is that only very few residues are allowed to lie outside the "favoured areas".
It has proven to be very difficult to improve the appearance of a Ramachandran plot while refining a structure. Therefore it is even more than 30 years after its design still useful as an a-posteriory quality check for a protein.
In the WHAT IF version of the plot, there are separate "favoured areas" for helix, strand, and other residues (secondary structure according to DSSP) represented in three different colours. In principle, all non glycine residues should lie in their correct area (since glycine does not have a side chain, it is not so restricted). In practice, a few residues (e.g. residues in active site loops) sometimes have strange backbone conformations, putting them outside the contoured areas. Hence it is very difficult to use the individual positions to judge local conformation. A better way to use the Ramachandran plot is by "overall impression"; this is something that we attempted to code in the Ramachandran Z-score: Negative is worse than average (WHAT IF will complain about values lower than -3, and complain strongly below -4), Positive is better than average. A bad example can be found in the report for "104L"
Impact on modelling by homology: If the Ramachandran plot and score are bad, the backbone of the structure is bad. Since model building by homology assumes the backbone is "correct", this will severely affect the quality of the model.
Impact for crystallographer: It is very difficult to use the results of a Ramachandran analysis in a refinement. Localized problems can warrant an extra look at the map. Remember that some outliers are expected in every structure. Global problems indicate low quality (low resolution?) data.
Omega angles are often fairly tightly restrained to 180 degrees. Too much so. According to statistics in small molecule structures, there is a fair amount of flexibility in omega angles.
WHAT IF will determine the variation in omega angles, and if it is very wide or very narrow it will complain.
Impact on modelling by homology: None.
Impact for crystallographer: Too tight restraints on omega could have a bad effect on the other backbone torsion angles phi and psi.
Different residue types in different secondary structure elements have different preferences for the chi-1/chi-2 rotamers. This preference can be treated in exactly the same way as the phi/psi torsion angles. WHAT IF will calculate a chi-1/chi-2 conformation Z-score.
A plot is not generated, as the preferences are not at all similar for different residue types (in phi/psi, 18 residues (all but GLY and PRO) are comparable).
Impact on modelling by homology: Quite heavy. If the existing side chain conformations are not correct, why bother to put the new ones in correctly.
Impact for crystallographer: Individual problems ask for a new look at the map: they could be indicative of disorder.
For the example, see "Rings and Planarity"
Nine of the amino acid side chains have a planar group (See J.Appl.Cryst. (1996) Vol 29 pp 714--716).
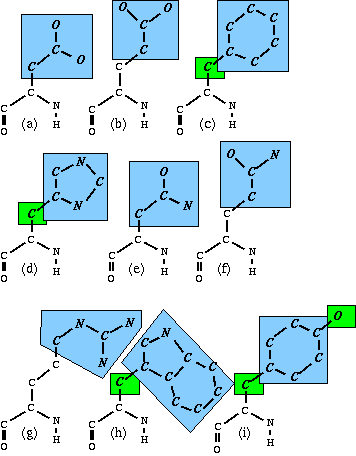
In this figure, the nine larger blue-shaded areas are the cores of the planar groups. There are 5 atoms in separate smaller green-shaded areas; these are in principle part of the shaded groups, but are treated separately.
Impact on modelling by homology: The offsets in atomic coordinates involved in planarity are normally quite low. But, if there are residues that have large planarity deviations, that could indicate strain in the structure that could be caused by incorrect backbone or side-chain positioning elsewhere. Be careful.
Impact for crystallographer: Planarity constraints in ASN, GLN, ASP and GLU in some programs are insufficient. Update to the latest version or complain with the author. If any problems remain: they could be an indication of a misplaced side chain (not necessarily this one, it could be another one pushing against it!).
Note: The planarity values for the groups shown in the picture above were determined 'the Engh and Huber way' from CSD data. For other planar groups such as nucleotides or peptide planes with protons attached, we have determined values that seemed reasonable. These values are not callibrated, and their check is thus called "uncallibrated planarity check".
Proline residues contain the only non-planar ring in protein structures. But the ring should not just be "non-planar": there are fairly nice rules about how far the deviation from planarity should be (the puckering amplitude), and which atom(s) should be out of the plane (the puckering phase).
If the puckering amplitude is too low, this could be an indication that the Proline residue is disordered over the two possible conformations. The "average" conformation is normally the result of the refinement, and this average ring is quite flat (see the report of "104L").
If the puckering amplitude is too high, or the puckering phase is way out of normal ranges, this could indicate a problem with the refinement. It could be an indication that the resolution is insufficient to study Proline puckering. It could also mean that the Proline residue is just "interesting".
The "normal" values used by the Proline puckering checks are subjective. They were assigned after a study of a large number of examples, but are no absolute boundaries. They should be handled with care.
Impact on modelling by homology: Minimal.
Impact for crystallographer: If a puckering amplitude is LOW, look in the map to see whether there is evidence for disorder. If the puckering phase is strange, look in the map whether you're sure this strange conformation is real.
In a globular protein, hydrophobic residues like phenylalanine like to sit "more on the inside", while hydrophilic residues like arginine like to sit "more on the outside" of the protein.
Obviously, a single exposed phenylalanine residue is not an indicator for a misthreaded structure; nor is a single buried arginine. Hence an inside/outside preference score for a single residue would never show anything unusual.
But: if a number of residues in sequence have strange locations, that could be an indication that e.g. a complete helix has been turned around, or (even worse) the complete structure is misthreaded. Or it could be an indication that the protein is not a normal, globular, water soluble protein.
WHAT IF will calculate an overall RMS Z-score to indicate how all residues in the protein are located. This value should be very close to 1.0 for globular proteins, and will be near 1.2 or larger for transmembrane proteins.
To make it easier to spot which areas in the protein are not feeling well, a sliding average plot is produced as well. Unusual areas in the structure will show up as peaks in this plot. At the top of the plot a secondary structure profile is shown to aid in the interpretation of the results.
Impact on modelling by homology: Difficult to say. In principle you don't want to model membrane proteins using the WHAT IF database, but since there is no alternatives. If a structure is really misfolded, it is better avoided as a template....
Impact for crystallographer: None? Just make sure there is an explanation for the strange behaviour...
For the example, see "Bumps"
Atoms can not overlap. Not without a reason, that is. The WHAT IF bump check will report excessive overlap between atoms that have no reason to be this close together.
Normal atom-pairs are allowed to "overlap" 0.4 Angstroms. (i.e. their inter-nuclear distance should be at least the sum of their Van der Waals radii minus 0.4 Angstrom).
Atoms are allowed to be fairly close together if they are bonded, or if there are only 2, 3 or (sometimes) 4 bonds between the pair. Another exception is a potential hydrogen bond connecting the two atoms. For all exceptions these exceptions, the bump criterium is relaxed by an appropriate amount.
All pairs that are too close together are listed in a table. They can be tagged at the end with codes like "HB" or "BF" or "B3", that indicate a potential reason for the overlap (but the reason is not good enough, otherwise the pair wouldn't be listed). The criteria applied are quite relaxed, so in principle there should be no bumps. Unfortunately, bumps appear to be rather common...
Impact on modelling by homology: For minor bumps minimal, but serious bumps could be an indicator of a serious problem with the structure.
Impact for crystallographer: Make sure there are no bumps. Bumps are physically unrealistic, they must be caused by a refinement problem, misplaced atoms, disorder, or forgotten "placeholders".
This will be obsolete soon: please check the "New packing quality" instead.
"packing quality" or "Directional atomic contact analysis" or "quality control" are three names for one of WHAT IF's older protein structure analysis tools. This analysis makes use of a so-called "threading"-potential: an empirical potential that expresses how well the sequence feels at home in the structure.
It would be possible to code such a check as a combination of "packing" facts, e.g.:
For each "fixed fragment" in a protein structure (any "largest group" of atoms that does not contain a torsion angle is a "fixed fragment") the occurrence of all possible atom types in all possible positions around the fixed fragment is counted. If a certain configuration occurs very frequently, it is assumed to be a preferred configuration. All preference counts for all atoms around a residue are used to calculate a summary score for each residue.
The advantage of a procedure like this is that all preferences are automatically coded; even preferences that are not known yet.
The new quality control score for each residue now is a Z-score that describes how well this residue feels compared to other similar residues in well refined structures. If the residue Z-score is negative, it feels less at home than the "average" residue. If the Z-score is positive, it feels more at home than average.
The individual scores are not very powerful. A lot of structures have a few low-scoring residues. More useful are the two other tables WHAT IF gives: a list of sequential residues that all have low scores (possibly indicating a mis-threaded segment), and the overall quality control Z-score.
Impact on modelling by homology: Severe. If a structure has a bad quality control Z-score, it can not be trusted.
Impact for crystallographer: The global quality control value should only be low for a really misthreaded structure. Individual residues listed are not really rare. The most interesting is the "residues in sequence" check: if that table shows any entries, have a look whether there is an alternative for the conformation of that "loop".
For the example see "Backbone"
Many of the checks in WHAT IF use the secondary structure codes given by DSSP to make a difference between residues in helices, extended conformations, and loops. In some cases, however, this split-up is insufficient: there could be large differences for residues at the beginning, in the middle or at the end of a helix. Or significant differences between different loop conformations.
In such cases WHAT IF uses complete context-dependency using a 3D backbone database. Here, a property of residue N is compared to database residues M for which the position of the C-alpha atoms of the residues N-2 throug N+2 are comparable to M-2 through M+2. This is slightly more context than just the phi and psi angles of the neighbouring residues, but a lot more flexible than using four sets of phi/psi. And it can be implemented in a very efficient way...
The checks using this context-dependent backbone database are described here.
The simplest check that can be performed tests the number of times a similar backbone conformation occurs in WHAT IF's database. If this is less than 3, the residue is listed as having a "unique" backbone conformation.
It is absolutely normal that this occurs a few times in a structure. But if the "Backbone normality" (described below) is low, the residues listed here might be a nice starting point for an extra study of the electron density.
Impact for crystallographer: Make sure that there is no obvious alternative conformation that is more normal.
The backbone normality is a structural average score that describes how well the backbone-database hits fit the structure.
The pepflip test looks at all the backbone hits for a residue, and then calculates the distance from the peptide-O in the query structure to each of the peptide-O's in the database hits. If most of the peptide hits in the database point in the opposite direction this could be an indication that the peptide plane should be flipped. All residues where this is found to be the case are listed.
This seems to happen sometimes for GLY residues, but even those are worth an extra look. Pepflips are also not uncommon for active site residues.
Obviously, the Pepflip analysis can only be done for residues for which the backbone conformation occurs a reasonable number of times in the database.
Impact on modelling by homology: It is difficult to fix a pepflip without the original X-ray data. If they are not very rare, extra care must be taken with the structure.
Impact for crystallographer: Make the pep-flip and look at loop conformation and hydrogen bonding patterns in the context of the electron density map. Choose the alternative that makes most sense.
The "rotamer check" is also a check based on the 3D backbone database in WHAT IF. For all the hits on the local backbone, the rotamer of "chi-1" is tabulated. At the end, it is verified whether the query residue falls in the most populated rotamer. If instead it falls in a minor rotamer class, for which only few examples are found in the database, the residue is tabulated.
Obviously, the rotamer check can only be done for residues for which the backbone conformation occurs a reasonable number of times in the database.
If a residue shows up here, there is quite a big chance that the "chi-1" rotamer is indeed incorrect. This affects the position of the complete side chain.
Impact for crystallographer: Make sure that there is no obvious alternative rotamer that is more normal.
For the example see "Water molecules".
The floating cluster analysis lists all water molecules that are not in contact with the protein structure, not even through other water molecules. Most likely, water molecules that show up here represent refinement artifacts.
Impact for crystallographer: Remove the listed water molecules.
Not a scientific problem but a technical problem is listed by the "symmetry relation" water check. The PDB wants to have all water molecules listed as close as possible to the protein molecule that is given in the PDB file. If any water molecules are signigicantly closer to symmetry related protein molecules, they are listed here together with suggested transformed coordinates.
Impact for crystallographer: Fix it.
For the example see "B-factors" and "B-factor plot".
"B-factors" or "temperature factors" are among the worst determined parameters in protein structures. In principle, they describe the flexibility of the atom positions in the structure. In effect, however, the B-factors are normally underdetermined, and hence absorb all kinds of inaccuracies in the X-ray structure.
The average B-factor for all atoms that have an accessibility value of 0 is calculated. This average is normally between 20 and 40. Lower values can be obtained for frozen crystals. Higher values are a possible indication of refinement problems.
WHAT IF also calculates what percentage of all atomic B-factors for non-accessible atoms is below 5.0. This should be very rare (less than 1%). Even in small molecule structures at room temperature such atoms are rare. If a lot of atoms have low B-factors, that is an indication that either the structure was determined using a frozen crystal, or there are significant refinement problems.
Since the B-factor is largely a parameter describing the motion of the protein atoms, and proteins are rather fixed entities that tend to rotate and translate as a whole, one expects the B-factors of atoms buried deep in the core to be lower than the ones at the surface of the protein. A bit less strict: one expects the B-factors of atoms that are close in space to be similar. Even less strict: one expects the B-factors of bonded atoms to be similar. And this is what WHAT IF's B-factor distribution check does: it verifies whether the difference of B-factors over all bonds is within expected ranges. The result is expressed as an RMS Z-score, which is expected to be around 1.0 for good structures.
The RMS Z-score for structures with 1 common B-factor per residue is around 0.35.
Dale Tronrud (J.Appl.Cryst. (1996) vol 29 pp 100-104) has described a very similar procedure to use during the refinement of protein structures. His calibration dataset has been much more carefully selected than ours, so in principle his analysis should be more correct. The problem is that he used structures of 1.6-1.8 Angstrom resolution as his calibration set, and he used only four of these. We suspect that the B-factors in these calibration structures have been moderately over-refined, and thus that his limits have not been set tight enough. Structures refined with Tronrud's method will show a "too-loose" error here, with a B-factor variability Z-score of around 1.8.
Impact on modelling by homology: Ignore this check for now. Many otherwise reliable structures have horrible B-factor distributions.
Impact for crystallographer: If the B-factors seem to go haywire, try to restrain them. Try to use Tronrud's procedure. If that is not an option use 2 B-factors per residue instead of 1 per atom: 1 for the backbone, and one for the side-chain.
For the example, see "Hydrogen bonds"
Protein crystallographers have difficulty to see 1 electron. Hence, it is not possible to see H atoms. But it is also fairly impossible in normal cases to distinguish "C" from "N" or "N" from "O".
The most visible effect of this problem is that ASN, GLN and HIS residues look symmetrical in the X-ray map, and often are placed the wrong way around.
By studying the hydrogen bond network around these residues, it is in most cases possible to distinguish between the alternatives. WHAT IF uses a sophisticated hydrogen bond force field specially designed for this purpose to check the positioning of the ASN, GLN and HIS side chains. It will list all of the residues that would have better hydrogen bonding if turned by 180 degrees.
Impact on modelling by homology: Make sure you turn the listed residues before starting the modelling.
Impact for crystallographer: Fix them, unless there is biological evidence or evidence from high-resolution B-factors or neutron diffraction that WHAT IF is wrong. WHAT IF normally isn't wrong.
As a side effect of the hydrogen bonding analysis, the protonation state of all HIS residues is determined as well, and all of them are listed here. The possibilities are:
Since hydrogen atoms are not given in most PDB files (and where they are, WHAT IF normally ignores them), WHAT IF can not know which protonation state was used in refinements. However, since there are slight bond-length differences between the three different rings, WHAT IF can "guess" which protonation state was used. Wherever it differs from the protonation state calculated using hydrogen bond analysis, it is printed in the table.
From this analysis it seems most people do not bother to put the correct HIS type in their structures: 80% of all recognizable HIS residues in the PDB database have been refined as HIS-H: the most unlikely of the three alternatives.
Impact on modelling by homology: None.
Impact for crystallographer: Make sure to use the right HIS type for the refinement.
For the example see "Summary for users of a structure".
For a quick impression of the whole structure, somebody that wants to use a structure for modelling or other structural studies can study the "summary for users". The numbers are identical to the numbers given earlier in the full report.
Impact on modelling by homology: In principle see the description of the individual numbers. But if there are a lot of "(poor)" or "(bad)" or "(loose)" qualifiers, you might want to avoid the structure altogether, without spending any more time on it.
Impact for crystallographer: None.
For the example see "Summary for depositors of a structure".
All the "structure average" numbers given in the check report are "absolute" quality indicators: an indication of the quality of the structure, independent of the way it was obtained. Those numbers are really nice for modellers, because it allows for easy comparison of structure quality. This makes it possible to select the "best of the two" structures for modelling.
For a depositor, however, the absolute quality of the structure is much less interesting. A depositor wants to know whether his 3.4 Angstrom structure is comparable in quality to other 3.4 Angstrom structures. The "summary for depositors" does this: it takes the verification results, and compares them with those obtained for other structures with similar resolution in the Protein Data Bank. The difference is expressed as a re-calibrated Z-score: -2.0 here means that a score is 2 sigma worse than the average structure of the same resolution, and +3.0 means this structure is a lot better than other structures in the same resolution class.
This recalibration is only performed for a-posteriori checks. The geometrical checks that do not depend on the X-ray data but only on the restraints used in the refinement are taken unchanged from the rest of the report.
Impact on modelling by homology: None.
Impact for crystallographer: Worry if any of the corrected numbers is below -3.0 (or all of them between -1.5 and -3.0, which also indicates a significant difference). Rejoice if any of the corrected numbers is above +3.0 (or all of them between +1.5 and +3.0, which also indicates a significant difference). Refer to the individual checks to find more information.